
Abstract
Dactylicapnos scandens (D. Don) Hutch (Papaveraceae) is a well-known traditional Chinese herb used for treatment of hypertension, inflammation, bleeding and pain for centuries. Although the major bioactive components in this herb are considered as isoquinoline alkaloids (IQAs), little is known about molecular basis of their biosynthesis. Here, we carried out transcriptomic analysis of roots, leaves and stems of D. scandens, and obtained a total of 96,741 unigenes. Based on gene expression and phylogenetic relationship, we proposed the biosynthetic pathways of isocorydine, corydine, glaucine and sinomenine, and identified 67 unigenes encoding enzymes potentially involved in biosynthesis of IQAs in D. scandens. High performance liquid chromatography analysis demonstrated that while isocorydine is the most abundant IQA in D. scandens, the last O-methylation biosynthesis step remains unclear. Further enzyme activity assay, for the first time, characterized a gene encoding O- methyltransferase (DsOMT), which catalyzes O-methylation at C7 of (S)-corytuberine to form isocorydine. We also identified candidate transcription factor genes belonging to WRKY and bHLH families that may be involved in the regulation of IQAs biosynthesis. Taken together, we first provided valuable genetic information for D. scandens, shedding light on candidate genes involved in IQA biosynthesis, which will be critical for further gene functional characterization.
Similar content being viewed by others

Introduction
Alkaloids are a large and diverse group of nitrogenous secondary metabolites that account for approximately 20% of plant species. Alkaloids mainly include isoquinoline alkaloids(IQAs), quinoline alkaloids, pyrrolidine alkaloids, and indole alkaloids, among which IQAs are the largest and most important alkaloids that specifically present in Magnoliaceae, Ranunculaceae, Papaveraceae and Berberidaceae. Current studies on IQAs with respect to separation of chemical constituents and pharmacodynamics functions showed that IQAs play key roles in anti-inflammatory and analgesia, thereby serving as the analgesics morphine and codeine, the antitumor agent noscapine, the muscle relaxant papaverine, and the antimicrobial agents sanguinarine and berberine1,2,3,4. However, the molecular mechanisms catalyzing and regulating IQAs biosynthesis in plants are still unclear because of the structural diversity of IQAs present in different plant species.
The biosynthesis of IQAs begins with conversion of L-tyrosine to dopamine and 4-hydroxyphenylacetaldehyde, which are then condensed to (S)-norcoclaurine by (S)-norcoclaurine synthase (NCS)5,6,7. Three methyltransferases [(S)-norcoclaurine 6-O-methyltransferase (6OMT), (S)-coclaurine-N-methyltransferase (CNMT) and (S)-3′-hydroxy-N-methylcoclaurine-4′-O-methyltransferase (4′OMT)] and one hydroxylase [(S)-N-methylcoclaurine 3′-hydroxylase (NMCH)] are involved in catalyzing the conversion of (S)-norcoclaurine to (S)-reticuline which has been considered as a common intermediate of many IQAs8,9,10,11. At present, biosynthetic pathways for several kinds of IQAs have been widely reported in a number of plant species, such as berberine in Coptis japonica, sanguinarine in Eschscholzia californica and morphine in Papaver somniferum, and many enzymes involved in IQAs biosynthesis in these plants have been characterized12,13,14. In the IQAs biosynthesis, many steps involve oxidation reaction catalyzed by cytochrome P450s (P450s) and O-methylation process catalyzed by OMTs family, which participate in synthesis of intermediate products (S)-reticuline, and subsequent multistep transformations to form different end products. P450 play a key role in oxidative reactions, including methylenedioxy bridge formation, intramolecular C–C phenol-coupling and intermolecular C–O phenol-coupling reactions15,16,17. CYP719B1 encoding salutaridine synthase in P. somniferum has been characterized to catalyze C-C phenol-coupling reaction in morphine biosynthesis18. Three CYP719A genes encoding canadine synthase, cheilanthifoline synthase and stylopine synthase, respectively, in C. japonica and E. californica catalyze methylenedioxy bridge-forming reactions in IQAs biosynthesis19, 20. Recently, it has been reported that (S)-cis-N-methylstylopine 14-hydroxylase (MSH), a member of the CYP82N subfamily of P450, catalyzes C–O couplings in sanguinarine biosynthesis from P. somniferum 21. O-methylation involves the transfer of the methyl group of SAM to the hydroxyl group of an acceptor molecule, resulting in formation of a methyl ether derivative and S-adenosyl-L-homocysteine. O-methylation of different C sites is catalyzed by different types of OMTs. Many OMTs in several plants have been characterized, including (1) 6OMT from C. japonica 8 and P. somniferum 22; (2) 4′OMT from C. japonica 8, P. somniferum 23, and E. californica 24; (3) norreticuline 7-O-methyltransferase (N7OMT) from P. somniferum 25; (4) reticuline 7-O-methyltransferase (7OMT) from P. somniferum 22; (5) scoulerine-9-O-methyltransferase (SOMT) from C. japonica 26 and P. somniferum 27; and (6) columbamine O-methyltransferase (CoOMT) from C. japonica 28. Recently, three O-methyltransferases, designated as SOMT1, SOMT2, and SOMT3 have been reported to be involved in noscapine biosynthesis in P. somniferum. SOMT1 is able to sequentially 9- and 2-O-methylate (S)-scoulerine, yielding (S)-tetrahydropalmatine, and also sequentially 3′- and 7-O-methylate both (S)-norreticuline and (S)-reticuline with relatively high substrate affinity, yielding (S)-tetrahydropapaverine and (S)-laudanosine, respectively. In contrast, SOMT2 and SOMT3 showed strict substrate specificity and regiospecificity as 9-O-methyltransferases targeting (S)-scoulerine27. Although IQAs biosynthesis such as morphine, sanguinarine and berberine has been well studied, other IQAs biosynthesis such as isocorydine and sinomenine is not well understood.
Dactylicapnos scandens (D. Don) Hutch, mainly distributing in northwestern India, Thailand, Tibet Autonomous Region and Yunnan Province in China, is a tuberous rooted perennial herb belonging to Papaveraceae29, 30. As a famous traditional Chinese medicine, it is a popular Bai folk medicine and has been used for treatment of inflammation, hypertension, bleeding and pain31. The main bioactive constituents of D. scandens are IQAs, including isocorydine, corydine, glaucine, sinomenine, protopine and magnoflorine32,33,34. Chemical investigation of D. scandens revealed that isocorydine was the most abundant34, 35, suggesting that it is a very good plant material to study the biosynthetic pathway of isocorydine. However, the genome of D. scandens has not been sequenced and genetic resources are scarce. In this study, we performed the transcriptomes of roots, leaves and stems of D. scandens using the Illumina HiSeq. 2000 sequencing platform. Based on previous reports12, 36,37,38 and current transcriptomic analysis, we proposed the integrated biosynthetic pathways for isocorydine, corydine, glaucine and sinomenine, and identified probably candidate genes that encode enzymes and transcription factors (TFs) controlling the IQAs biosynthesis in D. scandens. Furthermore, we characterized a candidate gene encoding OMT protein, DsOMT, which catalyzes O-methylation at C7 of (S)-corytuberine to form isocorydine.
Results and Discussion
Illumina sequencing and de novo assembly
To obtain a comprehensive understanding of D. scandens transcriptome, the cDNA libraries were constructed from total RNA of D. scandens roots, leaves and stems, respectively (Fig. 1). Three biological replications for each tissue were sequenced using the Illumina HiSeq. 2000 sequencing platform. After filtering out adaptor sequences, ambiguous reads and low-quality reads (Q20 < 20), a total of 3.7, 3.9 and 3.6 Gb of high-quality reads from roots, leaves and stems, respectively, were generated (Supplementary Table S1). The high-quality reads obtained in this study were deposited in the NCBI SRA database (accession number: SRA480383).

Roots, leaves and stems of D. scandens. Wild seedlings of D. scandens were transplanted into the field for 1.5 years.
Because genome information is unavailable for D. scandens, all clean reads were de novo assembled using Trinity software39, with optimized k-mer length of 25. We obtained a total of 96,741 unigenes with sequence length ranged from 201 to 17,943 bp, and a total length of 114,753,746 bp. The average length of all unigenes is 905 bp, and there are 34,151 unigenes (26.94%) longer than 1,000 bp. The coding DNA sequences (CDS) from all D. scandens unigene sequences were also detected and a total of 30,190 CDSs were obtained. Among them, 26.93% unigenes have CDS longer than 1,000 bp. The summary of sequencing and assembly results was shown in Table 1, and the length distribution of the transcripts, unigenes and CDS were shown in Supplementary Fig. S1.
Functional annotation
To annotate these assembled unigenes as many as possible, sequences were searched against seven public protein databases: NCBI nucleotide (NT), NCBI non-redundant protein (NR), SwissProt protein, Gene Ontology (GO), euKaryotic Ortholog Groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Protein family (PFAM). A total of 37,783 unigenes (39.05%) were annotated in the public databases and, of these, 5317 unigenes were annotated in all databases. There were 28,557 unigenes (29.51%) matched in the NR database, and 22,315 unigenes (23.06%) matched with known proteins in the SwissProt database. A total of 23,412 unigenes (24.20%) matched to the GO database and 11,545 unigenes (11.93%) matched to the KOG. The number of unigenes matched to the NT, KEGG and PFAM databases was 20,673 (21.36%), 10,516 (10.87%) and 23,149 (23.92%), respectively (Table 2).
For GO analyses, 415,147 unigenes were classified into three classes, including biological processes (274,720 unigenes), cellular components (86,318 unigenes), and molecular functions (54,109 unigenes) (Supplementary Fig. S2). There were 12,915 unigenes assigned to KOG classifications, which were divided into 25 specific categories. Predominantly unigenes were found in the category of general functional prediction that is associated with only basic physiological and metabolic functions (2,038, 15.78), whereas unigenes belonging to category of cell motility was the smallest group, with only four unigenes (0.03%) (Supplementary Fig. S3). The KEGG pathway-based analysis is helpful for understanding the biological functions and interactions of genes. A total of 10,782 unigenes had significant matches in the KEGG database and were assigned to 129 biological pathways. The category with the largest number of unigenes was metabolism that includes the biosynthesis of other secondary metabolites (367, 6.32%). Among them, predominantly category was phenylpropanoid biosynthesis (166 unigenes, 45.23%), followed by IQAs biosynthesis (44, 11.99%) and other biosynthesis (Supplementary Fig. S4).
Quantitative analysis of three major IQAs in D. scandens
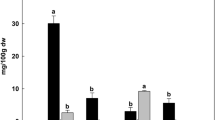
According to previous reports, isocorydine, sinomenine and protopine are the main IQA components of D. scandens 32,33,34. Herein, the content of these major IQAs in roots, leaves and stems of D. scandens was quantified by high performance liquid chromatography (HPLC). Compared to authentic standards, the contents of isocorydine, sinomenine and protopine in roots were 6.074, 0.822 and 1.824%, respectively (Fig. 2A,B), indicating that D. scandens roots are abundant in isocorydine. By contrast, the contents of isocorydine, sinomenine and protopine in leaves and stems were very low and almost undetectable (Fig. 2C, D). These results were in accordance with roots being the main medicinal part of the plant. Isocorydine, an aporphine alkaloid with one free hydroxyl group, can inhibit cell proliferation by inducing G2/M cell cycle arrest and apoptosis and target the drug-resistant cellular side population through PDCD4-related apoptosis in hepatocellular carcinoma (HCC)40,41,42, thus can be served as a potential antitumor agent in HCC. This indicates that isocorydine is a kind of pharmaceutically valuable IQAs. However, the molecular mechanisms involved in isocorydine biosynthesis remain unclear.

Chromatograms of three major IQAs in roots, leaves and stems of D. scandens. (A) HPLC chromatograms of sinomenine, isocorydine and protopine standards. HPLC chromatograms of sinomenine, isocorydine and protopine in D. scandens (B) roots, (C) leaves and (D) stems.
Candidate genes encoding enzymes involved in IQAs biosynthesis
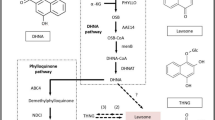
We focused on the discovery of genes involved in IQAs biosynthesis, which begins with the condensation of two L-tyrosine derivatives: 4-hydroxyphenylacetaldehyde and dopamine43, 44. Through a series of enzymatic reactions, (S)-reticuline is synthesized, which acts as the central intermediate and is diverted to different branches for biosynthesis of different types of IQAs45. In D. scandens, the major active constituents are isocorydine, corydine, glaucine, sinomenine, protopine and magnoflorine32,33,34. To date, although the protopine and magnoflorine pathways have been characterized in other plant sepcies17, 46, pathways of isocorydine, corydine, glaucine and sinomenine have not been determined. Therefore, we proposed their biosynthesis pathways based on previous reports and present transcriptome data (Fig. 3).

Putative pathways for IQAs biosynthesis in D. scandens. Enzymes found in this study are boxed. Abbreviations: TyrAT, L-tyrosine aminotransferase; 4HPPDC, 4-hydroxyphenylpuruvate decarboxylase; TYDC, tyrosine decarboxylase; 3OHase, tyrosine/tyramine 3-hydroxylase; NCS, (S)-norcoclaurine synthase; 6OMT, norcoclaurine 6-O-methyltransferase; CNMT, (S)-coclaurine N-methyltransferase; NMCH, N-methylcoclaurine 3′-hydroxylase; 4′OMT, 3′-hydroxy-N-methylcoclaurine 4′-O-methyltransferase; BBE, berberine bridge enzyme; CFS, (S)-cheilanthifoline synthase; SPS, (S)-stylopine synthase; TNMT, (S)-tetrahydroprotoberberine N-methyltransferase; MSH, (S)-cis-N-methylstylopine 14-hydroxylase; CTS, corytuberine synthase; SCNMT, (S)-corytuberine-N-methyltransferase; SalSyn, salutaridine synthase.
Based on the KEGG pathway assignment, we discovered the transcripts encoding all the known enzymes for (S)-reticuline biosynthesis from our Illumina dataset, which include L-tyrosine aminotransferase (TyrAT), tyrosine decarboxylase (TYDC), tyrosine/tyramine 3-hydroxylase (3OHase), NCS, 6OMT, CNMT, NMCH and 4′OMT. Protopine and magnoflorine biosynthesis pathways have previously been depicted as follows: (S)-scoulerine is formed from (S)-reticuline by berberine bridge enzyme (BBE)47, 48. The conversion of (S)-scoulerine to protopine begins with the formation of two methylenedioxy bridges by cheilanthifoline synthase (CFS) and stylopine synthase (SPS), both are members of the CYP719 family, forming (S)-stylopine. CFS and SPS have been isolated and characterized from E. californica 16, 49 and Mexican prickly poppy (Argemone mexicana)50. Subsequent N-methylation of (S)-stylopine by tetrahydroprotoberberine -N-methyltransferase (TNMT)51 yields (S)-N-methylstylopine, which is converted by (S)-cis-N-methylstylopine 14-hydroxylase (MSH) to protopine21. In magnoflorine biosynthesis, (S)-reticuline is first oxidized to (S)-corytuberine by (S)-corytuberine synthase (CTS) and, subsequently, (S)-corytuberine-N-methyltransferase (SCNMT) converts (S)-corytuberine to magnoflorine46 (Table 3; Supplementary Table 2; Fig. 3). Furthermore, 67 unigenes encoding enzymes involved in IQAs biosynthesis were used to detect their expression levels in different tissues based on reads per kilobase of transcript per million reads mapped (RPKM) values. The results indicated that most genes showed higher expression in roots than leaves or stems, especially for those genes located at downstream of protopine and isocorydine biosynthesis such as CFS, SPS, TNMT, MSH and CTS (Fig. 4). This is basically consistent with the higher content of IQAs in the roots of D. scandens (Fig. 2).

RPKM values of candidate unigenes involved in the biosynthesis of IQAs in D. scandens. Expression levels of candidate unigenes involved in the biosynthesis of IQAs in D. scandens leaves, stems and roots.
In the biosynthetic pathway of isocorydine, corydine and glaucine, we proposed that the last O-methylation reactions were catalyzed by OMTs by affecting different hydroxyl groups. In our dataset, we obtained 10 and 1 unigenes encoding 6OMT and 4′OMT, respectively. Unigenes c36937_g1, c77767_g1, DsOMT, c55366_g1, c32115_g1 and c53376_g1 were highly similar to P. somniferum PsOMT1, PsOMT2 and PsOMT3, respectively27. To characterize the evolutionary relationships between OMTs from D. scandens and known OMTs from other plant species, Neighbor-Joining tree was constructed. As shown in Fig. 5, unigenes c55366_g1 and c32115_g1 had high homology with 6OMT, while c53376_g1 had high homology with 4′OMT. Notably, we also found that c36937_g1, c77767_g1 and c47357_g1 were not clustered with the known OMTs, suggesting that they may have distinct roles in IAQs biosynthesis, which requires further study.

Phylogenetic tree of OMTs. Phylogenetic tree was constructed based on the deduced amino acid sequences for the D. scandens OMTs (bold letters) and other plant OMTs. Abbreviations and GenBank accession numbers for the sequences used are as follows: EcOMT, putative Eschscholzia californica OMT (ACO90220.1); CjCoOMT, Coptis japonica columbamine OMT (Q8H9A8.1); TtOMT, Thalictrum tuberosum catechol OMT (AAD29845.1); TtCaOMT, T. tuberosum catechol OMT (AAD29843.1); PsCaOMT, P. somniferum catechol OMT (AAQ01670.1); PsN7OMT, P. somniferum norreticuline 7OMT (ACN88562.1); Ps6OMT, P. somniferum norcoclaurine 6OMT (AAP45315.1); Ps4′OMT2, P. somniferum 3′-hydroxy-N-methylcoclaurine 4′OMT2 (AAP45314.1); Ps4′OMT1, P. somniferum 3′-hydroxy-N-methylcoclaurine 4′OMT1 (AAP45314.1); Cc4′OMT, Coptis chinensis 3′-hydroxy-N-methylcoclaurine 4′OMT (ABY75613.1); Cj4′OMT, C. japonica 3′-hydroxy-N-methylcoclaurine 4′OMT (Q9LEL5.1); Tf4′OMT, T. flavum 3′-hydroxy-N-methylcoclaurine 4′OMT (AAU20768.1); Cj6OMT, C. japonica norcoclaurine 6OMT (Q9LEL6.1); Tf6OMT, T. flavum norcoclaurine 6OMT (AAU20765.1); VvReOMT, Vitis vinifera resveratrol OMT (CAQ76879.1); PtFlOMT, Populus trichocarpa flavonoid OMT predicted protein (XP_002312933.1); CjSOMT, C. japonica scoulerine 9OMT (Q39522.1); TfSOMT, T. flavum scoulerine 9OMT(AAU20770.1); PaCafOMT, Picea abies caffeate OMT (CAI30878.1); CaCafOMT, Capsicum annuum caffeate OMT (AAG43822.1); PsOMT1, P. somniferum SOMT1 (JN185323); PsOMT2, P. somniferum SOMT2 (JN185324); PsOMT3, P. somniferum SOMT3 (JN185325); ObEuOMT, Ocimum basilicum eugenol OMT (AAL30424.1); MpFlOMT, Mentha X piperita flavonoid 8OMT (AAR09600.1); ObCafOMT, O. basilicum caffeate OMT (AAD38189.1); CbEuOMT, Clarkia breweri (iso)eugenol OMT (AAC01533.1); CbCafOMT, C. breweri caffeate OMT (O23760.1); and AmCafOMT, Ammi majus caffeate OMT (AAR24095.1).
Previous studies have confirmed the upstream biosynthesis pathway of isocorydine, and the corresponding enzymes have also been characterized36, 37. However, the last O-methylation step of isocorydine biosynthesis had not been elucidated yet. Here, we supplemented and perfected the isocorydine biosynthesis pathways because it is the most abundant in D. scandens (Fig. 2). By comparing molecular structure, we found isocorydine was formed by O-methylation at C7 on (S)-corytuberine. Thus, three functionally unknown OMT unigenes, c36937_g1, c77767_g1 and c47357_g1 (designated as DsOMT) were selected to study their function by in vitro enzyme activity. The results showed that DsOMT was able to O-methylate at C7 on (S)-corytuberine yielding isocorydine (Fig. 6). Therefore, we first established a complete biosynthesis pathway for isocorydine, which will be of potential significance for further understanding the molecular mechanisms of IQAs biosynthesis in plants.

HPLC analysis of O-methylation activity of recombinant DsOMT on (S)-corytuberine. (A) His-tag purified recombinant DsOMT on 12% SDS-PAGE gel, Lane M: Protein Marker, Lane 1: Un-purified, Lane 2: Flow through, Lane 3: Elution; (B) verified reaction equation; (C) (S)-corytuberine with denatured purified DsOMT proteins (control) and in vitro assay product of (S)-corytuberine with DsOMT; Product was identified using authentic standards, 1. (S)-corytuberine; 2. isocorydine.
Identification of transcription factors potentially involved in IQAs biosynthesis
Transcription factors (TFs) play a crucial role in secondary metabolism by regulating expression of related genes at the transcriptional level to control the flux of secondary metabolites. So far, researches on TFs regulating IQAs biosynthesis have mainly focused on WRKY and basic helix-loop-helix (bHLH) families. In C. japonica cells, suppressing the expression of CjWRKY1 and CjbHLH1 significantly decreased the expression of genes related to berberine biosynthesis52,53,54. Some Arabidopsis MYC2-type bHLH TF, such as NbbHLH1/NbbHLH2/NtMYC2 and CrMYC2, have been reported to be involved in the regulation of nicotine biosynthesis in Nicotiana plants and terpenoid indole alkaloid biosynthesis in Catharanthus roseus 55,56,57,58. Recently, Yamada et al. (2015) also reported two non-AtMYC2-type genes (EcbHLH1–1 and EcbHLH1-2) from E. californica were homologous to CjbHLH1, and the suppression of EcbHLH1 genes, particularly EcbHLH1-2, resulted in down-regulated expression of some IQA biosynthetic enzyme genes and sanguinarine accumulation59. In the transcriptomic data of D. scandens, we identified 71 unigenes encoding bHLH TFs, and 52 unigenes encoding WRKY TFs (Supplementary Table S3). The phylogenetic relationship between TFs from D. scandens and TFs characterized from other plants was showed in Fig. 7. We identified three unigenes (c51183_g3, c30931_g1, and c13730_g1) belonged to bHLH, and c51183_g3 was closely homologous to EcbHLH1-1. Six unigenes (c25015_g1, c46912_g1, c21995_g2, c50940_g4, c11393_g1, and c41587_g1) were clustered with WRKY1, with c41587_g1 highly homologous to CjWRKY1, suggesting that they might be involved in regulation of IQAs biosynthesis in D. scandens. Characterizing the functions of these unigenes will help us to better understand the regulatory mechanism of IQAs biosynthesis.

Phylogenetic analysis of TFs. Phylogenetic tree was constructed based on the deduced amino acid sequences for D. scandens TFs (bold letters) and other plant TFs involved in IQA biosynthesis. Accession Nos. are: Q39204, AtMYC2; Q8W2F1, AtMYC1; Q9ZVB5, AtbHLH100; AAQ14331, Catharanthus roseus CrMYC1; AAQ14332, CrMYC2; GQ859152, OsbHLH144; BAF14724, OsbHLH146; EEC73367, poplar PtbHLH130; EEE73911, Nicotiana benthamiana NbbHLH1; GQ859153, NbbHLH2; GQ859158, N. tabacum NtMYC1a; GQ859160, NtMYC2a; GQ859159, NtMYC1b; GQ859161, NtMYC2b; DT752478, Aquilegia formosa AfbHLH1; FD498024, Liriodendron tulipifera LtbHLH1; DT584473, Saruma henryi ShbHLH1; FD755492, Aristolochia fimbriata ArbHLH1; AB910896, EcbHLH1-1; AB910897, EcbHLH1-2; AB267401, CjWRKY1; AB564544, CjbHLH1; ADT82685, C. roseus CrWRKY1.
Conclusions
We firstly carried out transcriptomic analysis of D. scandens and obtained a total of 96,741 unigenes, which provide valuable genetic resource for this invaluable Chinese herb medicine. We further proposed the integrated biosynthetic pathways of isocorydine, corydine, glaucine and sinomenine in D. scandens. The identification of 67 unigenes and in vitro enzymatic characterization of one of them provide opportunities for the de novo production of active ingredients by microorganism engineering. In addition, identification and phylogenetic analysis of WRKY and bHLH TFs potentially involved in regulation of IQAs biosynthesis is of great importance to reveal molecular basis of IQAs biosynthesis pathways.
Methods
Plant materials
Wild seedlings of D. scandens collected from Weishan County, Yunnan Province, southwest China (25°23′N, 100°33′E and altitude: 2900 m) were transplanted into the field for 1.5 years. The roots, leaves and stems were harvested separately, immediately frozen in liquid nitrogen, and stored at −80 °C until use.
cDNA library construction, sequencing and de novo assembly
Total RNA was extracted from roots, leaves and stems using Trizol reagent (Invitrogen, New York, USA) following by purification with RNeasy MiniElute Cleanup Kit (Qiagen, Hilden, Germany), according to the manufacturers’ protocols. For mRNA library construction and deep sequencing, at least 20 μg of total RNA samples were prepared using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina sequencing on a Hiseq. 2000 platform. Three biological replications were performed for each tissue.
Raw reads were firstly transformed into clean reads by removing reads with sequencing adaptors, reads with frequency of unknown nucleotides above 5% and low-quality reads (containing more than 50% bases with Q-value ≤ 20) using a custom Perl script. Then, the clean reads were de novo assembled using the Trinity program (k-mer = 25, group pairs distance = 300) with default parameters43.
Functional annotation and candidate gene prediction
For functional annotations, all unigenes were assessed by public databases, including NT, NR (http://www.ncbi.nlm.nih.gov/), SwissProt (http://www.expasy.ch/sprot) and KOG database (http://www.ncbi.nlm.nih.gov/COG/), using BLASTX (E-value < 10−5) and BLASTN (E-value < 10−5), respectively. The unigenes were also aligned to KOG and KEGG databases (http://www.genome.jp/kegg)60 using BLASTX with an E-value < 10−10. A Perl script was used to retrieve KEGG Orthology information from blast result and then established pathway associations between unigenes and databases. Based on the results of the NR database annotation, the Blast2GO program61 was used to obtain GO unigene annotations. Then, WEGO62 software was used to perform GO classification and draw a GO tree. Moreover, the conserved domains/families of the assembled unigenes encoding proteins were searched against the Pfam database (version 26.0)63using Pfam_Scan script.
The CDSs of all unigenes were predicted using BLSATX and ESTscan. The unigene sequences were searched against the NR, KOG, KEGG and SwissProt protein databases using BLASTX (E-value < 10−5). The best alignment results were used to determine the sequence direction of unigenes. Unigenes with sequences with matches in one database were not searched further. When a unigene was not aligned to any database, ESTScan64 was used to predict coding regions and determine sequence direction. To identify the TFs, all unigenes were searched against the PlnTFDB database65 using iTAK analysis tool (http://bioinfo.bti.cornell.edu/cgi-bin/itak/index.cgi)66.
HPLC analysis
0.2 g dried powder of D. scandens roots, leaves and stems was respectively extracted with 50 mL of 1% hydrogen chloride −70% methanol mixed liquor for 60 min, and sonicated for 30 min. For determining main bioactive components of D. scandens, an Agilent 1260 HPLC system (Agilent Technologies, Santa Clara, CA, USA) was used. Chromatographic separation was performed on the chromatographic column Agilent Zorbar SB-C 18 (250 mm × 4.6 mm, 5 μm, Agilent Technologies) at a column temperature of 30 °C. The flow rate was fixed at 1 mL/min, and the mobile phase consisted of sodium dihydrogen phosphate-methanol (35:65, v/v) containing 0.1% sodium dodecyl sulphate (A) and acetonitrile (B). Separation was achieved using the following gradient system: 85% B at 0 min, 100% B at 10 min, and 100% B at 30 min. Detection was performed at 289 nm67. Authentic (S)-corytuberine, isocorydine, sinomenine and protopine were purchased from JK chemical (Beijing, China).
Digital gene expression profiling
The high-quality reads were aligned to the assembled unigenes with the BWA program68. An RPKM value was calculated for each unigene in each tissue of D. scandens. The RPKMs of all annotated isoforms for the same gene were summed as the RPKM of that gene. Differential expression of unigenes was calculated with a threshold of P value < 0.001 and two-fold change.
Phylogenetic analysis
Phylogenetic analysis was performed based on the deduced amino acid sequences of OMTs and TFs from D. scandens and other plants. All deduced amino acid sequences were aligned with Clustal X using the default parameters as described previously69: gap opening penalty, 10; gap extension penalty, 0.1; and delay divergent cutoff, 25%. The evolutionary distances were computed using MEGA5.10 with the Poisson correction method. For the phylogenetic analysis, a neighbor-joining tree was constructed using MEGA5.0. Bootstrap values obtained after 1000 replications are indicated on the branches. The scale represents 0.1 amino acid substitutions per site.
Recombinant protein purification and enzyme activity assay
Full-length cDNA of DsOMT was obtained by PCR amplification using primers 5′- CATATGATGAATCACAAAGTGCATCATCAT-3′ (forward, with the added NdeI restriction site underlined) and 5′-TCTAGATTATTTGCAGAACTCCATGACCCA-3′ (reverse, with the added XbaI restriction site underlined), and cloned into the pCzn1 vector (Zoonbio Biotechnology, China). The vector was introduced into the Escherichia coli line Arctic-Express (Zoonbio Biotechnology, China) for protein expression. The expression of the recombinant protein was induced by 0.5 mM of IPTG at 11 °C for 8 h. The cells were harvested by centrifugation and resuspended in binding buffer, and the suspension was subsequently homogenized by 1 h of 200Wsonication (Vibra Cell VC 505 Sonicator; Sonics & Materials, Newtown, CT). Cell debris was subsequently removed with 10-min centrifugation at 12,000 rpm. After renaturation by 2 M urea, the protein was purified by Ni-IDA -Sepharose CL-6B (Spectrum Chemical Manufacturing, USA) under the manufacturer’s instructions. The purity of the His-tagged protein was determined by SDS-PAGE followed by Coomassie Brilliant Blue staining.
The standard enzyme assay for DsOMT activity was performed using a reaction mixture in 50 μl of 100 mM Gly-NaOH (pH 9.0), 25 mM sodium ascorbate, 100 μM SAM, 10% (v/v) glycerol, 1 mM β-mercaptoethanol, 100 μM potential alkaloid substrate, and 50 μg of purified recombinant enzyme. Assays were carried out at 37 °C for 2 h and terminated by adding 200 μL of 1 M NaHCO3. Products were identified by HPLC as described above. Control was performed with denatured purified His-tagged proteins prepared by boiling in water for 20 min.
References
Holkova, I. et al. Involvement of lipoxygenase in elicitor-stimulated sanguinarine accumulation in Papaver somniferum suspension cultures. Plant Physiol Bioch. 48, 887–892 (2010).
Dang, T. T. T., Onoyovwi, A., Farrow, S. C. & Facchini, P. J. Biochemical genomics for gene discovery in benzylisoquinoline alkaloid biosynthesis in opium poppy and related species. Method Enzymol. 515, 231–266 (2012).
Facchini, P. J. Alkaloid biosynthesis in plants: Biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu Rev Plant Phys. 52, 29–66 (2001).
Kong, W. J. et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 10, 1344–1351 (2004).
Samanani, N. & Facchini, P. J. Purification and characterization of norcoclaurine synthase - The first committed enzyme in benzylisoquinoline alkaloid biosynthesis in plants. Biol Chem. 277, 33878–33883 (2002).
Samanani, N., Liscombe, D. K. & Facchini, P. J. Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J. 40, 302–313 (2004).
Minami, H., Dubouzet, E., Iwasa, K. & Sato, F. Functional analysis of norcoclaurine synthase in Coptis japonica. J Biol Chem. 282, 6274–6282 (2007).
Morishige, T., Tsujita, T., Yamada, Y. & Sato, F. Molecular characterization of the S-adenosyl-L-methionine:39-hydroxy-N-methylcoclaurine 4′- O-methyltransferase involved in isoquinoline alkaloid biosynthesis in Coptis japonica. J Biol Chem. 275, 23398–23405 (2000).
Choi, K. B., Morishige, T., Shitan, N., Yazaki, K. & Sato, F. Molecular cloning and characterization of coclaurine N-methyltransferase from cultured cells of Coptis japonica. J Biol Chem. 277, 830–835 (2002).
Sato, F., Inui, T. & Takemura, T. Metabolic engineering in isoquinoline alkaloid biosynthesis. Curr Pharm Biotechno. 8, 211–218 (2007).
Ziegler, J. & Facchini, P. J. Alkaloid biosynthesis: Metabolism and trafficking. Annu Rev Plant Biol. 59, 735–769 (2008).
Hagel, J. M. & Facchini, P. J. Benzylisoquinoline alkaloid metabolism–a century of discovery and a brave new world. Plant Cell Physiol. 54, 647–672 (2013).
Sato, F. Characterization of plant functions using cultured plant cells, and biotechnological applications. Biosci. Biotechnol. Biochem. 77, 1–9 (2013).
Morishige, T., Tamakoshi, M., Takemura, T. & Sato, F. Molecular characterization of O-methyltransferases involved in isoquinoline alkaloid biosynthesis in Coptis japonica. Proceedings of the Japan Academy, Series B. 86, 757–768 (2010).
Chapple, C. Molecular-genetic analysis of plant cytochrome P450-dependent monooxygenases. Annu Rev Plant Phys. 49, 311–343 (1998).
Mizutani, M. & Sato, F. Unusual P450 reactions in plant secondary metabolism. Arch Biochem Biophys. 507, 194–203 (2011).
Takemura, T., Ikezawa, N., Iwasa, K. & Sato, F. Molecular cloning and characterization of a cytochrome P450 in sanguinarine biosynthesis from Eschscholzia californica cells. Phytochemistry. 91, 100–108 (2013).
Gesell, A. et al. CYP719B1 is salutaridine synthase, the CC phenol-coupling enzyme of morphine biosynthesis in opium poppy. Journal of Biological Chemistry. 284, 24432–24442 (2009).
Ikezawa, N. et al. Molecular cloning and characterization of CYP719, a methylenedioxy bridge-forming enzyme that belongs to a novel P450 family, from cultured Coptis japonica cells. J Biol Chem. 278, 38557–38565 (2003).
Ikezawa, N., Iwasa, K. & Sato, F. Molecular cloning and characterization of methylenedioxy bridge-forming enzymes involved in stylopine biosynthesis in Eschscholzia californica. FEBS J. 274, 1019–1035 (2007).
Beaudoin, G. A. W. & Facchini, P. J. Isolation and characterization of a cDNA encoding (S)-cis-N-methylstylopine 14-hydroxylase from opium poppy, a key enzyme in sanguinarine biosynthesis. Biochem Biophys Res Commun. 431, 597–603 (2013).
Ounaroon, A., Decker, G., Schmidt, J., Lottspeich, F. & Kutchan, T. M. R,S)-Reticuline 7-O-methyltransferase and (R,S)-norcoclaurine 6-O-methyltransferase of Papaver somniferum - cDNA cloning and characterization of methyl transfer enzymes of alkaloid biosynthesis in opium poppy. Plant J. 36, 808–819 (2003).
Ziegler, J., Diaz-Chávez, M. L., Kramell, R., Ammer, C. & Kutchan, T. M. Comparative macroarray analysis of morphine containing Papaver somniferum and eight morphine free Papaver species identifies an Omethyltransferase involved in benzylisoquinoline biosynthesis. Planta. 222, 458–471 (2005).
Inui, T., Tamura, K., Fujii, N., Morishige, T. & Sato, F. Overexpression of Coptis japonica norcoclaurine 6-O-methyltransferase overcomes the ratelimiting step in benzylisoquinoline alkaloid biosynthesis in cultured Eschscholzia californica. Plant Cell Physiol. 48, 252–262 (2007).
Pienkny, S., Brandt, W., Schmidt, J., Kramell, R. & Ziegler, J. Functional characterization of a novel benzylisoquinoline O-methyltransferase suggests its involvement in papaverine biosynthesis in opium poppy (Papaver somniferum L). Plant J. 60, 56–67 (2009).
Takeshita, N. et al. Molecular cloning and characterization of S-adenosyl-L-methionine-scoulerine-9-O-methyltransferase from cultured cells of Coptis japonica. Plant Cell Physiol. 36, 29–36 (1995).
Dang, T. T. & Facchini, P. J. Characterization of three O-methyltransferases involved in noscapine biosynthesis in opium poppy. Plant Physiol. 159, 618–631 (2012).
Morishige, T., Dubouzet, E., Choi, K. B., Yazaki, K. & Sato, F. Molecular cloning of columbamine O-methyltransferase from cultured Coptis japonica cells. Eur J Biochem. 269, 5659–5667 (2002).
The National Traditional Chinese Medicines Compile Group, The National Traditional Chinese Medicines Compilation. The Peoples Medicine Publishing House, Beijing, p. 856 (1996).
Frisby, S. & Hind, N. 622. dactylicapnos scandens. Curtis’s Bot. Mag. 25, 207–215 (2008).
State Administration of Traditional Chinese Medicine of the Peoples Republic of China, Zhonghua Bencao, Shanghai, Shanghai Press of Science and Technology 3, p. 641 (1999).
Wu, Y., Zhao, Y., Liu, Y. & Zhou, J. Alkaloids from Dactylicapnos scandens. Natural Product Research & Development 20, 622–626 (2008).
Wang, F. et al. Alkaloids from Dactylicapnos scandens Hutch. Zhongguo Zhong yao za zhi 34, 2057–2059 (2009).
Wang, X. et al. Preparative isolation of alkaloids from Dactylicapnos scandens using pH-zone-refining counter-current chromatography by changing the length of the separation column. Journal of chromatography. B. Analytical technologies in the biomedical and life sciences 879, 3767–3770 (2011).
Guo, C. et al. Application of a liquid chromatography-tandem mass spectrometry method to the pharmacokinetics, tissue distribution and excretion studies of Dactylicapnos scandens in rats. J Pharmaceut Biomed. 74, 92–100 (2013).
Beaudoin, G. A. W. & Facchini, P. J. Benzylisoquinoline alkaloid biosynthesis in opium poppy. Planta 240, 19–32 (2014).
Hagel, J. M. et al. Metabolome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Biol. 15, 220 (2015).
Chang, L., Hagel, J. M. & Facchini, P. J. Isolation and Characterization of O-methyltransferases Involved in the Biosynthesis of Glaucine in Glaucium flavum. Plant Physiol. 169, 1127–1140 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011).
Sun, H. et al. Isocorydine inhibits cell proliferation in hepatocellular carcinoma cell lines by inducing G2/m cell cycle arrest and apoptosis. PLoS One. 7, 36808 (2012).
Lu, P. et al. Isocorydine targets the drug-resistant cellular side population through PDCD4-related apoptosis in hepatocellular carcinoma. Mol Med. 18, 1136–1146 (2012).
Chen, L. et al. Derivate isocorydine inhibits cell proliferation in hepatocellular carcinoma cell lines by inducing G2/M cell cycle arrest and apoptosis. Tumour Biol. 37, 5951–5961 (2016).
Liscombe, D. K., MacLeod, B. P., Loukanina, N., Nandi, O. I. & Facchini, P. J. Evidence for the monophyletic evolution of benzylisoquinoline alkaloid biosynthesis in angiosperms. Phytochemistry 66, 1374–1393 (2005).
Lee, E. J. & Facchini, P. Norcoclaurine synthase is a member of the pathogenesis-Related 10/Bet v1 protein family. Plant Cell. 22, 3489–3503 (2010).
Agarwal, P. et al. Comparative analysis of transcription factor gene families from Papaver somniferum: identification of regulatory factors involved in benzylisoquinoline alkaloid biosynthesis. Protoplasma 253, 857–871 (2016).
Ikezawa, N., Iwasa, K. & Sato, F. Molecular cloning and characterization of CYP80G2, a cytochrome P450 that catalyzes an intramolecular C-C phenol coupling of (S)-reticuline in magnoflorine biosynthesis, from cultured Coptis japonica cells. J Biol chem. 283, 8810–8821 (2008).
Facchini, P. J., Penzes, C., Johnson, A. G. & Bull, D. Molecular characterization of berberine bridge enzyme genes from opium poppy. Plant Physiol. 112, 1669–1677 (1996).
Winkler, A., Hartner, F., Kutchan, T. M., Glieder, A. & Macheroux, P. Biochemical evidence that berberine bridge enzyme belongs to a novel family of flavoproteins containing a bi-covalently attached FAD cofactor. J Biol Chem. 281, 21276–21285 (2006).
Ikezawa, N., Iwasa, K. & Sato, F. CYP719A subfamily of cytochrome P450 oxygenases and isoquinoline alkaloid biosynthesis in Eschscholzia californica. Plant Cell Rep. 28, 123–133 (2009).
Chavez, M. L. D., Rolf, M., Gesell, A. & Kutchan, T. M. Characterization of two methylenedioxy bridge-forming cytochrome P450-dependent enzymes of alkaloid formation in the Mexican prickly poppy Argemone mexicana. Arch Biochem Biophys. 507, 186–193 (2011).
Liscombe, D. K. & Facchini, P. J. Molecular cloning and characterization of tetrahydroprotoberberine cis-N-methyltransferase, an enzyme involved in alkaloid biosynthesis in opium poppy. J Biol Chem. 282, 14741–14751 (2007).
Kato, N. et al. Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant cell physiol. 48, 8–18 (2007).
Apuya, N. R. et al. Enhancement of alkaloid production in opium and California poppy by transactivation using heterologous regulatory factors. Plant Biotechnol J. 6, 160–175 (2008).
Yamada, Y. et al. Isoquinoline alkaloid biosynthesis is regulated by a unique bHLH-type transcription factor in Coptis japonica. Plant cell physiol. 52, 1131–1141 (2011).
Todd, A. T., Liu, E. W., Polvi, S. L., Pammett, R. T. & Page, J. E. A functional genomics screen identifies diverse transcription factors that regulate alkaloid biosynthesis in Nicotiana benthamiana. Plant J. 62, 589–600 (2010).
Shoji, T. & Hashimoto, T. Tobacco MYC2 regulates jmonate-inducible nicotine biosynthesis genes directly and by way of the NIC2-locus ERF Genes. Plant Cell Physiol. 52, 1117–1130 (2011).
Zhang, H. T. et al. The basic helix-loop-helix transcription factor CrMYC2 controls the jasmonate-responsive expression of the ORCA genes that regulate alkaloid biosynthesis in Catharanthus roseus. Plant J. 67, 61–71 (2011).
Zhang, H. B., Bokowiec, M. T., Rushton, P. J., Han, S. C. & Timko, M. P. Tobacco transcription factors NtMYC2a and NtMYC2b form nuclear complexes with the NtJAZ1 repressor and regulate multiple jasmonate-inducible steps in nicotine biosynthesis. Mol Plant. 5, 73–84 (2012).
Yamada, Y., Motomura, Y. & Sato, F. CjbHLH1 homologs regulate sanguinarine biosynthesis in Eschscholzia californica cells. Plant cell physiol. 56, 1019–1030 (2015).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, 354–357 (2006).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, 293–297 (2006).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, 222–230 (2014).
Iseli, C., Jongeneel, C. V. & Bucher, P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 99, 138–148 (1999).
Perez-Rodriguez, P. et al. PInTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, 822–827 (2010).
Dai, X. B., Sinharoy, S., Udvardi, M. & Zhao, P. X. PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics 14, 321 (2013).
Zhang, G. H. et al. Transcriptome analysis of Panax vietnamensis var. fuscidicus discovers putative ocotillol-type ginsenosides biosynthesis genes and genetic markers. BMC Genomics 16, 159 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Jiang, N. H. et al. Analysis of the transcriptome of Erigeron breviscapus uncovers putative scutellarin and chlorogenic acids biosynthetic genes and genetic markers. Plos One 9, e100357 (2014).
Acknowledgements
This work was supported by the pilot project for establishing new socialized service system by agricultural science and education combination in Yunnan Province (Medical Plant Unit) (2014NG003); the project of young and middle-aged talent of Yunnan Province (2014HB011).
Author information
Authors and Affiliations
Contributions
W.F. and S.Y. conceived the study. S.H., W.S., K.C., X.W., Y. D., J.C., J.Z., and G.Z. performed the experiments and carried out the analysis. S.H., W.F., S.Y., Y.D. G.Z., and J.Y. designed the experiments and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, S.M., Song, W.L., Cong, K. et al. Identification of candidate genes involved in isoquinoline alkaloids biosynthesis in Dactylicapnos scandens by transcriptome analysis. Sci Rep 7, 9119 (2017). https://doi.org/10.1038/s41598-017-08672-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08672-w
This article is cited by
-
Ethnomedicine, phytochemistry, and pharmacological activities of Uvaria chamae P. Beauv.: A comprehensive review
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
The chromosome-level reference genome of Coptis chinensis provides insights into genomic evolution and berberine biosynthesis
Horticulture Research (2021)
-
Full-length transcriptome analysis of Coptis deltoidea and identification of putative genes involved in benzylisoquinoline alkaloids biosynthesis based on combined sequencing platforms
Plant Molecular Biology (2020)
-
Identification and characterization of methyltransferases involved in benzylisoquinoline alkaloids biosynthesis from Stephania intermedia
Biotechnology Letters (2020)
-
Transcriptome analysis of Panax zingiberensis identifies genes encoding oleanolic acid glucuronosyltransferase involved in the biosynthesis of oleanane-type ginsenosides
Planta (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
