
Abstract
Honeybee (Apis mellifera) health is threatened globally by the complex interaction of multiple stressors, including the parasitic mite Varroa destructor and a number of pathogenic viruses. Australia provides a unique opportunity to study this pathogenic viral landscape in the absence of V. destructor. We analysed 1,240A. mellifera colonies across Australia by reverse transcription-polymerase chain reaction (RT-PCR) and next-generation sequencing (NGS). Five viruses were prevalent: black queen cell virus (BQCV), sacbrood virus (SBV), Israeli acute paralysis virus (IAPV) and the Lake Sinai viruses (LSV1 and LSV2), of which the latter three were detected for the first time in Australia. We also showed several viruses were absent in our sampling, including deformed wing virus (DWV) and slow bee paralysis virus (SBPV). Our findings highlight that viruses can be highly prevalent in A. mellifera populations independently of V. destructor. Placing these results in an international context, our results support the hypothesis that the co-pathogenic interaction of V. destructor and DWV is a key driver of increased colony losses, but additional stressors such as pesticides, poor nutrition, etc. may enable more severe and frequent colony losses to occur.
Similar content being viewed by others


Introduction
Increased awareness of the importance of pollination for functional ecosystems and global food security has seen a surge in research into pollinator health and population declines1, 2. Unravelling the complexity of increased honeybee (Apis mellifera) colony losses has been a particular focus of current research because of the economic value of honey production and crop pollination. Recent research implicates multiple stressors, including parasites, pathogens, chemicals, poor nutrition, climate and beekeeper management3,4,5,6. Arguably the most significant of these factors is the parasitic mite, Varroa destructor, which feeds on developing honeybee larvae and adult bees and transmits lethal viruses7,8,9,10,11. Since shifting host from the Asian honeybee (A. cerana) to A. mellifera in the mid-20th century, this mite has spread worldwide causing significant honeybee losses12. The emergence of V. destructor has also significantly altered the viral landscape in honeybee populations globally by increasing virus transmission and causing selection of more virulent virus strains13,14,15,16. The combination of V. destructor and viruses is now considered the major cause of global colony losses11, 13, 17, 18, but uncovering the importance of viruses alone remains a significant challenge due to the ubiquitous presence of V. destructor.
Viruses typically persist as covert infections in honeybee populations, but outbreaks occur when colonies become stressed or encounter certain environmental conditions19. The expansion of V. destructor has significantly increased colony stress and elevated the importance of viruses in colony losses11, 13, 17, 18. This has led to increased understanding of the role of pathogenic viruses within colonies and identified deformed wing virus (DWV) as a major co-pathogen involved in colony losses in association with V. destructor 17, 18, 20,21,22. Although DWV was only discovered following the spread of V. destructor, it is thought to exist naturally at low prevalence in all honeybee populations14, 23. This hypothesis was recently tested following the arrival of V. destructor in Hawaii14 and New Zealand24, showing that DWV prevalence increased while strain diversity decreased. However, data for historical virus prevalence before V. destructor introduction is rare and typically predates the discovery of DWV25, 26.
There are now only a handful of A. mellifera populations in the world that are not infested by V. destructor. Most of these are small island populations with small-scale beekeepers. Australia is the exception by having a large commercial-scale beekeeping industry that is exposed to similar agrochemical and pathogen stressors as North America and Europe, but remains free of V. destructor. Australia has also not experienced the increased colony losses reported overseas. Australia’s mite-free status therefore provides an excellent stage for comparing viral landscapes with and without the confounding effects of V. destructor, hence providing insights into the causes of global colony losses. The last extensive studies of honeybee viruses in Australia relied on serological methods to examine apiaries during 1980 and 198326, 27. This identified the presence of sacbrood virus (SBV), black queen cell virus (BQCV), Kashmir bee virus (KBV), chronic bee paralysis virus (CBPV) and cloudy wing virus (CWV). Since then honeybee virology in Australia has continued sporadically28, 29, but remains under-researched and molecular screening for viruses has been non-existent.
With the help of molecular methods it is possible to expand our knowledge of the diversity of viruses in Australia. Of particular note is the apparent absence of the DWV complex (DWV-A, DWV-B and DWV-C)15, 22, 30. There are no reported symptoms (e.g. wing deformities, shortened abdomens), but there is speculation that Australian honeybees have covert infections of DWV and recent overseas studies claim to have detected DWV-A in imported bee samples31, 32. There are also other viruses linked with V. destructor including acute bee paralysis viruses (ABPV), Israeli acute paralysis virus (IAPV) and slow bee paralysis virus (SBPV) that have not been tested for in Australia using molecular methods. Furthermore, several new viruses such as the Lake Sinai viruses (LSV1 and LSV2) have been recently identified overseas using next-generation sequencing (NGS)33, of which there is no information for in Australia. NGS technology has been applied in multiple insect systems for virus discovery33,34,35,36,37,38 and presents a valuable tool for characterising the viral landscape of Australian honeybees.
In this paper, we characterised the Australian honeybee viral landscape by both direct detection using reverse-transcription PCR (RT-PCR) and a NGS approach to deliver wider coverage and sensitivity. We hypothesised that there would be lower virus prevalence in Australian honeybees and DWV would be absent, compared to honeybee populations in the presence of V. destructor. Our results showed that in the absence of V. destructor there was still a significant prevalence and diversity of honeybee viruses, but this did not include the DWV complex or several other viruses that have been linked to V. destructor pathogenicity. The implications of these results for unravelling the complex epidemiology of global honeybee colony losses are discussed.
Results
Virus prevalence in Australian apiaries
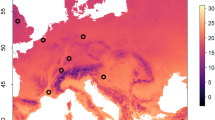
Honeybees were analysed from 1,240 hives representing 155 independent apiaries across five chosen regions (Fig. 1). Five honeybee viruses were detected in adult bee samples, with BQCV being the most common virus (65%) followed by LSV1 (45%), SBV (35%), LSV2 (27%) and IAPV (21%). There was some consistency across locations and seasons, although LSV1, SBV and LSV2 were at higher prevalence in several locations (Fig. 1, Table 1). In Region 2, LSV1 and SBV had equal highest prevalence in March 2014, while LSV1 was the most common virus in Region 1 and the only virus detected in Region 4, although no viruses were found in 3 KUN samples that formed part of Region 4. In Region 5, where there are fewer managed colonies, LSV2 had highest prevalence. IAPV was notably rare outside Region 1 and 3, with only one positive sample in Region 2. An additional 27 brood samples from across all regions that were suspected of virus infection tested positive for SBV.

Map of Australia showing sampling sites (●) and apiary origins (▲) across five regions (1–5) and prevalence of five honeybee viruses detected by RT-PCR in adult honeybee samples. Samples for Region 1 include: a, VIC/NSW-1 August 2013; b, VIC/NSW-2 April 2014; c, VIC/NSW-3 August 2014; d, SA August 2014; e, QLD August 2013; f, QLD October 2014. Samples for Region 2 include g, WA-1 October 2013; h, WA-2 March 2014. Region 3–5 were comprised of single sampling periods. Map was created using DIVA-GIS version 7.5 (www.diva-gis.org).
Overall, virus infections were very common in Australian honeybees with only 14% of samples free of the target viruses. Multiple viruses were detected in the majority of samples (61%), with four viruses co-infecting between 3% and 17% of samples in Regions 1 and 2 and all five viruses detected in one Region 1 sample (SA). However, five other honeybee viruses were not detected in any region; DWV, SBPV, ABPV, KBV and CBPV, which included an additional 124 brood samples tested for DWV and SBPV.
NGS of Australian honeybee populations
NGS was used to gain a more complete picture of the honeybee virus landscape in Australia by identifying virus genomes in nine pooled adult bee samples from Regions 1–4. This approach increased our sensitivity for detecting low prevalence viruses and avoided potential problems with primer specificity. All five viruses detected by RT-PCR were identified in each pooled sample, with a high number of reads covering the full genome in most cases. Normalised read counts also provided an indication of relative infection levels for each virus (Table 2). Consistent with the prevalence data, BQCV was commonly the most abundant virus with SBV and IAPV having high read counts in some samples. LSV1 and LSV2 were generally least abundant suggesting few overt infections from these viruses. Interestingly, NGS revealed all five viruses in the NT sample when only LSV1 was detected by RT-PCR. LSV1 had the highest read count (490,875 reads) while the other viruses had approximately 30,000 reads between them.
A small number of sequence reads mapped to the DWV and SBPV and CBPV reference genomes when using a lower similarity threshold of 0.7 (Table 2). Manual inspection of these reads revealed only short sequence fragments (<200 nucleotides) aligned to the reference genomes with variable sequence similarity (Supplementary material 2). For example, 15 sequence fragments aligned to all three DWV strains with an overall similarity of 69–74% (Supplementary material 3). This level of identity from only limited sequence fragments is insufficient to support the presence of these viruses, despite both DWV and SBPV existing as virus complexes with 16–20% variation22, 39. A number of sequence reads also mapped to the KBV reference genome, but still had higher identity with IAPV.
Australia’s honeybees are unique on the colony stress spectrum
A comparison of 41 similar virus studies highlighted the unique stress profile of Australia’s honeybee population (Supplementary material 4). No other A. mellifera population has been identified that has confirmed the absence of V. destructor and DWV, and also has a large commercial-scale beekeeping industry. We distilled these studies into five stress profiles representing the spectrum of global A. mellifera populations to show that V. destructor and DWV are key factors in increased colony losses (Table 3).
Diversity of honeybee viruses
Phylogenetic analysis of consensus genomes revealed considerable diversity between sampling regions and Australian isolates typically formed distinct clades from overseas isolates. BQCV isolates from this study were split into two distinct clades, where the winter isolates from VIC/NSW and SA grouped separately from the remaining Australian isolates (Fig. 2a). Similarly, SBV isolates formed several distinct clusters, with those from VIC/NSW and SA having greater similarity to a Korean isolate from A. mellifera than the original UK reference strain (Fig. 2b).

Maximum likelihood consensus tree of (a) SBV isolates based on 2,761 amino acids where AC indicates isolates obtained from A. cerana and (b) BQCV isolates based on 2,412 amino acids. Isolates from this study are indicated by (●).
IAPV has been implicated in colony losses in Israel and the USA, and Australian honeybees have been suggested as the source population. Interestingly, only the SA isolate grouped closely with an overseas strain, which was a USA isolate obtained from imported Australian bees (Fig. 3a). Remaining Australian isolates were split into two clades. The VIC/NSW-1 and TAS isolates were positioned among USA isolates, while the other isolates branched together as an outside clade. Therefore despite potential pathways through trade of live bees, this result does not indicate that Australian honeybees are the source population for IAPV.

Maximum likelihood consensus tree of (a) IAPV isolates based on 2,849 amino acids and (b) LSV isolates based on 600 amino acids of the RdRP gene. Isolates of IAPV, LSV1 and LSV2 from this study are indicated by (●) and variant LSV isolates from this study are indicated by (▲). The NT isolates were excluded because of low genome coverage.
LSV1 and LSV2 are recently described viruses from the USA that have since been detected in Europe and have been also correlated with colony losses. The SA and TAS isolates were most similar to the original USA strains, while the remaining isolates separated into several distinct groups with some geographic clustering (Fig. 3b). De novo assembly also revealed the presence of multiple variant LSV strains. Partial genomes were recovered from all samples that shared approximately 70% identity to LSV1 and LSV2. Other LSV strains (LSV3–LSV7) with similar levels of divergence have also been identified from Europe and the USA40,41,42 and from two previous Australian studies43, 44. Only partial gene sequences are available for these variants, but high similarity was found between the QLD, VIC/NSW-1 and VIC/NSW-3 variants with Cairns isolates (94–97%) and the NT variant was 99% and 97% similar to LSV5 and LSV3 sequences. Phylogenetic analysis of the RNA-dependent RNA polymerase (RdRp) gene showed several of the variant LSV isolates formed a separate clade related to LSV1 isolates. We propose that this clade be consolidated with related LSV strains (e.g. LSV3, LSV5) and designated LSV3. The WA-2 and VIC/NSW-2 variant LSVs were distinct from all currently known LSV strains and 86% similar to each other. We propose these variants be designated to a new LSV group, LSV8.
Discussion
This study provides the first comprehensive molecular-based analysis of Australia’s honeybee viral landscape and highlighted key contrasts with overseas landscapes affected by V. destructor. We found five honeybee viruses (BQCV, LSV1, SBV, IAPV and LSV2) that were common and, in contrast to our hypothesis, virus prevalence was still high despite the absence of V. destructor. Virus prevalence in this study was comparable with, or higher, than similar studies in Europe11, 45,46,47,48,49,50,51, North America33, 52, 53 and Asia54,55,56. Differences in study design make direct comparisons difficult, but our data support that viruses can achieve high prevalence without V. destructor acting as a mechanical and biological vector. It is well established that honeybee viruses can persist as covert infections in colonies and high apiary level prevalence does not necessarily equate to high infection levels within hives57, but using NGS data as an indication of abundance suggests all five viruses were present at levels greater than expected for covert infections. It is uncertain how indicative these levels are of pre-Varroa levels or if other pathogens have induced high virus prevalence in Australia. The microsporidian parasites Nosema apis and N. ceranae have reported synergistic relationships with BQCV and potentially other viruses and could be driving the high virus prevalence in Australia58, 59. Interestingly BQCV in this study was highest in the winter samples (August), which coincides with peak Nosema spp. infections60, and the winter BQCV isolates formed a distinct clade. Seasonality will certainly influence virus prevalence, such that longitudinal monitoring of viruses in Australia would further add to our understanding of viral dynamics in the absence of Varroa and DWV.
We have provided strong evidence that several viruses are not present in Australian honeybees. Of particular interest is the apparent absence of the DWV complex, which is suggested to exist at low levels in all honeybee populations without V. destructor 14. While it is fundamentally difficult to prove complete absence of DWV, we have taken a weight of evidence approach that is consistent with the World Organisation for Animal Health (OIE) guidelines to support this conclusion. This approach not only considers the negative detection of DWV in our structured RT-PCR survey, but also includes the results of our sensitive NGS analysis, the lack of clinical signs of DWV-associated disease, limited pathways for virus introduction and the previous lack of detection in imported Australian bees by the USA using a NGS analysis52. Collectively, this evidence gives strong support for the absence of DWV in Australia. This finding contradicts two recent overseas studies that have reportedly detected DWV-A in acquired Australian bee samples31, 32. In both studies, short sequences were recovered by quantitative PCR from a small sample set and these showed high similarity (>98%) to local DWV-A strains. Based on our results we believe these detections were likely false positives or contamination. It seems improbable for DWV to be detected in such limited samples and not detected in this wider study using equally sensitive methods. However, our NGS analysis did map several short sequence fragments (<200 nucleotides) to the DWV reference strains under low stringency conditions. While some of these fragments were within the accepted variation of the DWV complex (up to 20%), these sequences overall were significantly different (69–74%) to each DWV strain (Supplementary material 2 and 3). Therefore it is most likely that these short sequences correspond to an undescribed distinct virus (or viruses) that is only distantly related to DWV.
Not detecting DWV in Australian A. mellifera also presents alternative evidence that DWV may have been absent from other populations before the arrival of V. destructor. Martin et al.14 demonstrated how the arrival of V. destructor on some Hawaiian islands and not others had resulted in the predominance of a single DWV strain. Their study is cited as evidence that DWV exists at low levels in all populations. However, Hawaii is not a completely closed population from the United States mainland and it is possible the V. destructor-free islands have had DWV introduced more recently through imported queens. A similar scenario may explain the presence of DWV on the V. destructor-free island of Newfoundland, Canada61. Despite import regulations, DWV may have been introduced through imported queens. There is also the possibility of contamination as samples were transferred to the United States for virus testing. Mondet et al.24 also provided an excellent study of viral dynamics in New Zealand at the V. destructor expansion front to show the emergence of DWV several years after the mite established. Again, it was uncertain whether DWV was present in New Zealand prior to the arrival of mites. Mondet et al.24 did not detect DWV beyond the expansion front and earlier virus testing did not detect DWV in New Zealand62, 63, suggesting a possible post-V. destructor introduction of DWV. Australia clearly provides an excellent environment to further examine the viral landscape before the potential arrival of V. destructor, which has been difficult to fully achieve elsewhere64. Although there have been some potential pathways for DWV introduction into Australia, e.g. imported queens, pest incursions, our results suggest that this virus complex has not established.
Research into the causes of bee declines observed in Europe and the USA illustrate that this is a complex problem with individual studies often leading to different conclusions. The current consensus is increased colony losses experienced in some countries are the result of multiple, interacting stresses3, 65. As it is very difficult to replicate these complex interactions experimentally, there is an important role for “natural experiments” and scenarios where only some of the identified potential causes of colony losses are present. Our study has confirmed that Australia’s A. mellifera population does not have either V. destructor or DWV, but it does harbour a high prevalence of other honeybee viruses as well as common fungal and bacterial diseases. In addition, Australian beekeepers have not reported repeated problems with increased colony losses of the scale seen in the Northern Hemisphere, despite being exposed to many of the same environmental stressors found overseas. Therefore, the Australian population offers a unique stage for comparative studies that can start to uncouple the importance of other stressors.
To fully interpret the Australian situation in the international context, and to graphically summarise the studies we compiled as part of our review (Supplementary material 4), we propose that colony stress might best be represented as a “spectrum” of severity, with Australia at one extreme with only sporadic colony losses and the other extreme being severe and repeated colony losses as reported in the USA (Fig. 4). We acknowledge that colony losses are not always unambiguous or reported in an unbiased or systematic way. Nonetheless, using this spectrum concept, we hypothesise that the co-pathogenic role of V. destructor and DWV is likely the primary underlying driver that interacts with variable environmental stressors, particularly other pathogens, agrochemicals and nutritional stress. The role of V. destructor and DWV as a key component in adversely affecting A. mellifera colony health is well established8, 10, 11. However, the emergence of increased colony losses in the Northern Hemisphere has led to other potential stressors such as neonicotinoid insecticides and novel pathogens to draw focus. Our analysis of Australian A. mellifera colonies shows that the presence of viruses other than DWV, even at high levels and combined with environmental stressors, appears to not have created increased colony losses. Therefore, we suggest that V. destructor and DWV is likely still the primary interaction. We note that other authors have made similar conclusions based on analysing the effect of V. destructor on colony health in Europe17, and thus our hypothesis is not entirely novel. However, by delivering new data from a V. destructor-free landscape, we have provided insight to enable a re-focus to the V. destructor – DWV co-pathogenic interaction.

A conceptualization of A. mellifera colony stress as a spectrum with the current Australian situation being at one end and experiencing only sporadic colony losses and the increased colony losses of the USA and parts of Europe at the right-most extent of the spectrum. We hypothesize that a key underlying driver creating this spectrum may be V. destructor and DWV acting as co-pathogens, and interacting with other pathogens and abiotic stressors. Most countries are situated in the middle, with variable occurrence and severity of colony losses.
Our study has also given the first direct confirmation of IAPV in Australia after being detected in the USA in imported Australian bees and being linked with Colony Collapse Disorder (CCD)52. High read abundance in Australian A. mellifera also suggests potential impacts on hive health, but in contrast there are no reports of increased colony losses or CCD. IAPV has now been shown to form a virus complex with KBV and ABPV66 and so earlier serological detections of KBV in Australia may have been IAPV67. If so, then IAPV is most likely a serotype of KBV, as Bailey et al.68 detected several serotypes of KBV in Australia. This idea is also supported by the absence of KBV in this study, whereas it was commonly detected in previous serological-based Australian surveys. In addition, IAPV was largely restricted to eastern Australia, a very similar distribution to that of KBV in previous serological-based surveys, which suggests either a more recent history for IAPV in Australia or that IAPV was previously regarded a serotype of KBV. This confusion highlights the issue of molecular-based virus identification without reference to previous identities based on serology. Interestingly, the presence of IAPV (and most likely KBV) and absence of ABPV in this population also supports a theory that these viruses occupy the same ecological niche, which leads to one virus dominating under different conditions69. For instance ABPV dominates in Europe, while IAPV dominates in Asia, Australia and the USA and KBV dominates in New Zealand. IAPV is still considered a serious pathogen of honeybees because of its high virulence, its occasional cause of natural honey bee mortality without an apparent synergist, and evidence that V. destructor is an effective biological vector70. However, the comparable prevalence of IAPV in Australia with overseas studies supports the hypothesis that, as a fast replicating virus, IAPV is not closely associated with V. destructor pathogenicity. Any A. mellifera brood killed by IAPV vectored by V. destructor, will die before emergence, thus preventing V. destructor offspring from leaving the capped brood cell18. Therefore, while V. destructor is capable of vectoring IAPV, the prevalence and abundance of this virus is most likely influenced more by other factors.
LSV1 and LSV2 are also new detections for Australia and extend the distribution of this virus complex. These viruses were identified as common viruses in the United States in 2009 and since then a diversity of LSV strains have been identified in Europe and North America33, 40, 41. With the detection of diverse Australian LSV strains it is apparent these viruses have had a long association with A. mellifera globally. In fact, a LSV variant was identified in a closed A. mellifera population on Norfolk Island, which has only had introductions of Australian honeybee stock44. In this study, LSV1 and LSV2 had consistently lower read abundance than the other detected honeybee viruses, suggesting a more stable coexistence with A. mellifera. In contrast, LSV2 has been recorded as the most abundant virus in US studies and associated with weak and collapsed colonies41, 42. There is much to learn about this diverse group and further characterisation of LSV variants will help focus efforts on strains relevant to honeybee health. Furthermore, if LSV1 and LSV2 have long existed in the global honeybee population, it is surprising they have only recently been detected. Perhaps both viruses were identified previously using serological methods, but were renamed after being molecularly identified, as appears to be the case with IAPV and KBV. It has been noted that bee virus X (BVX) and bee virus Y (BVY), share multiple similarities with LSV1 and LSV2 but have not yet been genetically characterised71. Further comparison of the physical characteristics of LSV1 and LSV2 with BVX and BVY is warranted.
This study has provided valuable insight into the honeybee viral landscape in the absence of V. destructor and delivered the first molecular analysis of honeybee viruses in Australia. It is clear from these findings that viruses can occur at high prevalence and abundance without the assistance of V. destructor mites and there is considerable viral diversity in this population. The absence of several viruses, DWV and SBPV in particular, is valuable biosecurity knowledge for Australia but additionally provides a unique opportunity for future comparative studies. Furthermore, it leads on to a testable hypothesis that V. destructor acting as a co-pathogen with DWV and interacting with environmental stressors is most likely to be the underlying driver of increased colony losses. Further examination and monitoring of the unique Australian A. mellifera population should aid our understanding of viral dynamics and pathogen interactions without the confounding effects of V. destructor or DWV and will contribute to global efforts to improve honeybee health.
Methods
Sampling strategy and collection
One-off samples from 155 apiaries totaling 1,240 hives were collected between August 2013 and April 2015 from five distinct regions in Australia (Fig. 1). Region 1 included Queensland (QLD), New South Wales (NSW), Victoria (VIC) and South Australia (SA), Region 2 was south Western Australia (WA), Region 3 was Tasmania (TAS), Region 4 was the Northern Territory (NT) and Kununurra (KUN) in north WA and Region 5 was Kangaroo Island (KI) off the SA coast. Eight seemingly healthy hives were randomly sampled at each apiary visited with two brood frames inspected from each hive to collect suspected diseased brood. Approximately 25 adult bees per hive were collected from the brood comb. An additional 124 brood samples suspected of viral disease (i.e. visually unhealthy but symptoms not consistent with bacterial or fungal disease) were collected separately to specifically test for DWV, SBPV and SBV. All samples were collected on ice, transported frozen to the laboratory and then stored at −20 °C.
RT-PCR analysis for honeybee viruses
Adult bee samples was split into four sub-samples of 50 bees and homogenized in 5 mL of 0.05 M potassium phosphate buffer. 500 μl of homogenate was cleared by adding 50 μl of diethyl ether and 100 μl of chloroform, vigorously shaking for 30 seconds and then centrifuging for 2 minutes at 7,000 g. The supernatant was collected and RNA extracted using the Purelink viral RNA extraction kit (Invitrogen) and cDNA generated using the Tetro cDNA synthesis kit (Bioline) following the manufacturer’s protocol using both random hexamer (50 ng/μl) and oligo (dT)18 (270 ng/μl) primers and RT incubation at 40 °C for 40 min. Each sample was tested for 10 honeybee viruses; sacbrood virus (SBV), black queen cell virus (BQCV), Kashmir bee virus (KBV), Israeli acute paralysis virus (IAPV), acute bee paralysis virus (ABPV), chronic bee paralysis virus (CBPV), deformed wing virus (DWV-A and DWV-B), slow bee paralysis virus (SBPV), Lake Sinai virus 1 (LSV1) and Lake Sinai virus 2 (LSV2). PCR assays were carried out in 10 μl reactions containing 1 x PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.4 μM forward primer, 0.4 μM reverse primer, 1 U Taq DNA polymerase (New England Biolabs) and 1 μl cDNA template. PCR cycling conditions were 30 cycles of 95 °C (15 s), 56 °C or 58 °C (30 sec), 72 °C (40 s) and primers are given in Supplementary material 1. PCR reactions were analysed on 1.2% agarose gels stained with GelRed (Jomar Biosciences) and all positive PCR products were confirmed by Sanger sequencing.
RNA preparation and library construction for NGS
Nine pooled adult bee samples from the nine sampling periods (VIC/NSW-1 – August 2013, VIC/NSW-2 – April 2014, VIC/NSW-3 – August 2014, SA – August 2014, QLD – October 2014, WA-1 – October 2013, WA-2 – October 2014, TAS – November 2014, NT – June 2014) were chosen for NGS. Pooled samples were created from 1 ml of 50 homogenized bees per apiary sample into a screw-cap centrifuge tube and adjusting with 0.05 M potassium phosphate buffer for a total volume of 20 ml. 3 ml diethyl ether and 3 ml chloroform were added, shaken vigorously and centrifuged at 6,000 rpm for 30 minutes (J-E Avanti centrifuge). Supernatants were transferred to Ultraclear SW28 tubes (Beckman Coultier) and centrifuged at 21,500 rpm for 3.5 hours at 4 °C (Beckman L-80 ultracentrifuge). Pelleted samples were dissolved in 1 ml of 0.05 M potassium phosphate buffer then passed through a 0.22 μm filter to remove bacterial contamination. We then mixed 340 μl of each filtered sample with 10 μl of RNase, 10 μl DNase and 40 μl of DNase I buffer and incubated at 37 °C for 2 hours. RNA was extracted from the treated samples using the Purelink viral RNA extraction kit. Illumina libraries were prepared using the TruSeq stranded mRNA library prep kit (Illumina, San Diego, CA) and 100 bp paired-end sequences were generated on an Illumina HiSeq. 2500 run in rapid mode at the Biomolecular Research Facility (Australian National University, Canberra). An average of 93 million paired-end reads were generated per library. Bioinformatics analysis of RNA-seq data was carried out with the CLC Genomic workbench (Qiagen, Aarhus). Raw data were quality trimmed and adapter sequences removed before the trimmed reads were mapped to reference virus genomes from the NCBI GenBank database of the honeybee viruses tested for by RT-PCR. Mapping parameters were first set with a length fraction of 0.9 and similarity fraction of 0.9, and a second mapping was run with the similarity fraction set to 0.7. Consensus sequences were manually inspected for genome coverage and similarity to mapped reference genomes using BLASTn. Trimmed data were also de novo assembled in CLC Genomics workbench using default parameters and a minimum contig length of 1,000 nucleotides. Contigs were compared against the NCBI non-redundant protein database using BLASTx and contigs of interest were inspected manually to detect potential variant genomes. Phylogenetic analysis of viral genomes were done in MEGA772. Mulitple amino acid sequence alignments with relevant reference sequences were performed with MUSCLE and consensus maximum-likelihood phylogenetic trees were constructed using appropriate models for each virus and 1,000 bootstraps.
Semi-systematic review of international bee virus studies
To place our results within the international context and the current debates on the causes of increased honeybee colony losses, we undertook a semi-systematic review of the literature to find comparable virus studies. Studies that presented prevalence data for multiple viruses across large areas or in relation to colony losses were included. The results from these studies were extracted into a summary table (Supplementary material 4).
References
Klein, A. M. et al. Importance of pollinators in changing landscapes for world crops. Proc. R. Soc. B-Biol. Sci. 274, 303–313, doi:10.1098/rspb.2006.3721 (2007).
Vanbergen, A. J. et al. Threats to an ecosystem service: pressures on pollinators. Front. Ecol. Environ. 11, 251–259, doi:10.1890/120126 (2013).
Goulson, D., Nicholls, E., Botias, C., & Rotheray, E. L. Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science 347, doi:10.1126/science.1255957 (2015).
Genersch, E. Honey bee pathology: current threats to honey bees and beekeeping. Appl. Microbiol. Biotech. 87, 87–97, doi:10.1007/s00253-010-2573-8 (2010).
Blacquiere, T., Smagghe, G., van Gestel, C. A. M. & Mommaerts, V. Neonicotinoids in bees: a review on concentrations, side-effects and risk assessment. Ecotoxicol. 21, 973–992, doi:10.1007/s10646-012-0863-x (2012).
Brodschneider, R. & Crailsheim, K. Nutrition and health in honey bees. Apidologie 41, 278–294, doi:10.1051/apido/2010012 (2010).
Rosenkranz, P., Aumeier, P. & Ziegelmann, B. Biology and control of Varroa destructor. J. Invert. Path. 103, S96–S119, doi:10.1016/j.jip.2009.07.016 (2010).
Guzmán-Novoa, E. et al. Varroa destructor is the main culprit for the death and reduced populations of overwintered honey bee (Apis mellifera) colonies in Ontario, Canada. Apidologie 41, 443–450 (2010).
Dahle, B. The role of Varroa destructor for honey bee colony losses in Norway. J. Apic. Res. 49, 124–125, doi:10.3896/ibra.1.49.1.26 (2010).
Dainat, B., Evans, J. D., Chen, Y. P., Gauthier, L., & Neumann, P. Predictive Markers of Honey Bee Colony Collapse. PloS One 7, doi:10.1371/journal.pone.0032151 (2012).
Genersch, E. et al. The German bee monitoring project: a long term study to understand periodically high winter losses of honey bee colonies. Apidologie 41, 332–352, doi:10.1051/apido/2010014 (2010).
Anderson, D. L. & Trueman, J. W. H. Varroa jacobsoni (Acari: Varroidae) is more than one species. Exp. Appl. Acarol. 24, 165–189 (2000).
Genersch, E., & Aubert, M. Emerging and re-emerging viruses of the honey bee (Apis mellifera L.). Vet. Res. 41, doi:10.1051/vetres/2010027 (2010).
Martin, S. J. et al. Global honey bee viral landscape altered by a parasitic mite. Science 336, 1304–1306, doi:10.1126/science.1220941 (2012).
McMahon, D. P. et al. Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R Soc. B-Biol. Sci. 283, doi:10.1098/rspb.2016.0811 (2016).
Moore, J. et al. Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. J. Gen. Virol. 92, 156–161, doi:10.1099/vir.0.025965-0 (2011).
Martin, S. J., Ball, B. V. & Carreck, N. L. The role of deformed wing virus in the initial collapse of varroa infested honey bee colonies in the UK. J. Apic. Res. 52, 251–258, doi:10.3896/IBRA.1.52.5.12 (2013).
McMenamin, A. J. & Genersch, E. Honey bee colony losses and associated viruses. Curr. Opin. Insect Sci. 8, 121–129, doi:10.1016/j.cois.2015.01.015 (2015).
Chen, Y. P., & Siede, R. In Advances in Virus Research Vol. 70, 33–80 (Elsevier Academic Press Inc, 2007).
Allen, M. & Ball, B. The incidence and world distribution of honey bee viruses. Bee World 77, 141–162 (1996).
Di Prisco, G. et al. A mutualistic symbiosis between a parasitic mite and a pathogenic virus undermines honey bee immunity and health. Proc. Natl. Acad. Sci. USA 113, 3203–3208, doi:10.1073/pnas.1523515113 (2016).
de Miranda, J. R. & Genersch, E. Deformed wing virus. J. Invert. Path. 103, S48–S61, doi:10.1016/j.jip.2009.06.012 (2010).
Bailey, L. & Ball, B. V. Honey bee pathology, 193 pp (1991).
Mondet, F., de Miranda, J. R., Kretzschmar, A., Le Conte, Y. & Mercer, A. R. On the Front Line: Quantitative Virus Dynamics in Honeybee (Apis mellifera L.) Colonies along a New Expansion Front of the Parasite Varroa destructor. PloS Pathog. 10, doi:10.1371/journal.ppat.1004323 (2014).
Ball, B. V. & Allen, M. F. The prevalence of pathogens in honey bee (Apis mellifera) colonies infested with the parasitic mite Varroa jacobsoni. Ann. Appl. Biol. 113, 237–244, doi:10.1111/j.1744-7348.1988.tb03300.x (1988).
Hornitzky, M. A. Z. Prevalence of virus infections of honeybees in eastern Australia. J. Apic. Res. 26, 181–185 (1987).
Anderson, D. L. & Gibbs, A. Viruses and Australian native bees. Austral. Beekeeper 83, 131–134 (1982).
Roberts, J. M. K. & Anderson, D. L. A novel strain of sacbrood virus of interest to world apiculture. J. Invert. Path. 118, 71–74, doi:10.1016/j.jip.2014.03.001 (2014).
Anderson, D. L. & Giacon, H. Reduced pollen collection by honeybee (Hymenoptera, Apidae) colonies infected with Nosema apis and Sacbrood Virus. J. Econ. Entomol. 85, 47–51 (1992).
Mordecai, G. J., Wilfert, L., Martin, S. J., Jones, I. M. & Schroeder, D. C. Diversity in a honey bee pathogen: first report of a third master variant of the Deformed Wing Virus quasispecies. ISME J. 10, 1264–1273, doi:10.1038/ismej.2015.178 (2016).
Wilfert, L. et al. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 351, 594–597, doi:10.1126/science.aac9976 (2016).
Singh, R. et al. RNA Viruses in Hymenopteran Pollinators: Evidence of Inter-Taxa Virus Transmission via Pollen and Potential Impact on Non-Apis Hymenopteran Species. PloS One 5, doi:10.1371/journal.pone.0014357 (2010).
Runckel, C. et al. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema and Crithidia. PLoS One 6, e20656 (2011).
Temmam, S. et al. Characterization of Viral Communities of Biting Midges and Identification of Novel Thogotovirus Species and Rhabdovirus Genus. Viruses-Basel 8, doi:10.3390/v8030077 (2016).
Ng, T. F. F. et al. Broad Surveys of DNA Viral Diversity Obtained through Viral Metagenomics of Mosquitoes. PloS One 6, doi:10.1371/journal.pone.0020579 (2011).
Nouri, S., Salem, N., Nigg, J. C. & Falk, B. W. Diverse Array of New Viral Sequences Identified in Worldwide Populations of the Asian Citrus Psyllid (Diaphorina citri) Using Viral Metagenomics. J. Virol. 90, 2434–2445, doi:10.1128/jvi.02793-15 (2016).
Granberg, F. et al. Metegenomic detection of viral pathogens in Spanish honeybees: co-infection by aphid lethal paralysis, Israel acute paralysis and Lake Sinai viruses. PLoS One 8, e57459 (2013).
Mordecai, G. J. et al. Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 6, doi:10.1038/srep34983 (2016).
de Miranda, J. R. et al. Genetic characterization of slow bee paralysis virus of the honeybee (Apis mellifera L.). J. Gen. Virol. 91, 2524–2530, doi:10.1099/vir.0.022434-0 (2010).
Ravoet, J., De Smet, L., Wenseleers, T. & de Graaf, D. C. Genome sequence heterogeneity of Lake Sinai Virus found in honey bees and Orf1/RdRP-based polymorphisms in a single host. Virus Res. 201, 67–72, doi:10.1016/j.virusres.2015.02.019 (2015).
Daughenbaugh, K. F. et al. Honey Bee Infecting Lake Sinai Viruses. Viruses-Basel 7, 3285–3309, doi:10.3390/v7062772 (2015).
Cornman, R. S. et al. Pathogen webs in collapsing honey bee colonies. PloS One 7, doi:10.1371/journal.pone.0043562 (2012).
Roberts, J. M. K. & Anderson, D. L. Establishing the disease status of the Asian honeybee in the Cairns region. Rural Industries Research and Development Corporation report 13/082, 1–28 (2013).
Malfroy, S. F., Roberts, J. M. K., Perrone, S., Maynard, G. & Nadine, C. A pest and disease survey of the isolated Norfolk Island honey bee (Apis mellifera) population. J.Apic. Res. 55(2), 202–211, doi:10.1080/00218839.2016.1189676 (2016).
Berenyi, O., Bakonyi, T., Derakhshifar, I., Koglberger, H. & Nowotny, N. Occurrence of six honeybee viruses in diseased Austrian apiaries. App. Environ. Microbiol. 72, 2414–2420, doi:10.1128/aem.72.4.2414-2420.2006 (2006).
Forgach, P., Bakonyi, T., Tapaszti, Z., Nowotny, N. & Rusvai, M. Prevalence of pathogenic bee viruses in Hungarian apiaries: Situation before joining the European Union. J. Invert. Path. 98, 235–238, doi:10.1016/j.jip.2007.11.002 (2008).
Tentcheva, D. et al. Prevalence and seasonal variations of six bee viruses in Apis mellifera L. and Varroa destructor mite populations in France. Appl. Environ. Microbiol. 70, 7185–7191, doi:10.1128/aem.70.12.7185-7191.2004 (2004).
Berthoud, H., Imdorf, A., Haueter, M., Radloff, S. & Neumann, P. Virus infections and winter losses of honey bee colonies (Apis mellifera). J. Apic. Res. 49, 60–65, doi:10.3896/ibra.1.49.1.08 (2010).
Nielsen, S. L., Nicolaisen, M. & Kryger, P. Incidence of acute bee paralysis virus, black queen cell virus, chronic bee paralysis virus, deformed wing virus, Kashmir bee virus and sacbrood virus in honey bees (Apis mellifera) in Denmark. Apidologie 39, 310–314, doi:10.1051/apido:2008007 (2008).
Ravoet, J. et al. Comprehensive bee pathogen screening in Belgium reveals Crithidia mellificae as a new contributory factor to winter mortality. PloS One 8, doi:10.1371/journal.pone.0072443 (2013).
Budge, G. E. et al. Pathogens as Predictors of Honey Bee Colony Strength in England and Wales. PloS One 10, doi:10.1371/journal.pone.0133228 (2015).
Cox-Foster, D. L. et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318, 283–287, doi:10.1126/science.1146498 (2007).
Desai, S. D., Kumar, S. & Currie, R. W. Occurrence, detection, and quantification of economically important viruses in healthy and unhealthy honey bee (Hymenoptera: Apidae) colonies in Canada. Can. Entomol. 148, 22–35, doi:10.4039/tce.2015.23 (2016).
Kojima, Y. et al. Infestation of Japanese Native Honey Bees by Tracheal Mite and Virus from Non-native European Honey Bees in Japan. Microb. Ecol. 62, 895–906, doi:10.1007/s00248-011-9947-z (2011).
Sanpa, S. & Chantawannakul, P. Survey of six bee viruses using RT-PCR in Northern Thailand. J. Invert. Path. 100, 116–119, doi:10.1016/j.jip.2008.11.010 (2009).
Tsevegmid, K., Neumann, P., & Yanez, O. The Honey Bee Pathosphere of Mongolia: European Viruses in Central Asia. PloS One 11, doi:10.1371/journal.pone.0151164 (2016).
Amiri, E., Meixner, M., Nielsen, S. L. & Kryger, P. Four Categories of Viral Infection Describe the Health Status of Honey Bee Colonies. PloS One 10, doi:10.1371/journal.pone.0140272 (2015).
Bailey, L., Ball, B. V. & Perry, J. N. Association of viruses with two protozoal pathogens of the honey bee. Ann. Appl. Biol. 103, 13–20, doi:10.1111/j.1744-7348.1983.tb02735.x (1983).
Doublet, V., Labarussias, M., de Miranda, J. R., Moritz, R. F. A. & Paxton, R. J. Bees under stress: sublethal doses of a neonicotinoid pesticide and pathogens interact to elevate honey bee mortality across the life cycle. Environ. Microbiol. 17, 969–983, doi:10.1111/1462-2920.12426 (2015).
Hornitzky, M. A study of Nosema ceranae in honeybees in Australia. Rural Industries Research and Development Corporation report 11/045, 1–37 (2011).
Shutler, D. et al. Honey Bee Apis mellifera Parasites in the Absence of Nosema ceranae Fungi and Varroa destructor Mites. PloS One 9, doi:10.1371/journal.pone.0098599 (2014).
Todd, J. H., De Miranda, J. R. & Ball, B. V. Incidence and molecular characterization of viruses found in dying New Zealand honey bee (Apis mellifera) colonies infested with Varroa destructor. Apidologie 38, 354–367, doi:10.1051/apido:2007021 (2007).
Berenyi, O. et al. Phylogenetic analysis of deformed wing virus genotypes from diverse geographic origins indicates recent global distribution of the virus. Appl. Environ. Microbiol. 73, 3605–3611, doi:10.1128/aem.00696-07 (2007).
Iwasaki, J. M., Barratt, B. I., Lord, J. M., Mercer, A. R. & Dickinson, K. J. The New Zealand experience of varroa invasion highlights research opportunities for Australia. Ambio, doi:10.1007/s13280-015-0679-z (2015).
Horn, J., Becher, M. A., Kennedy, P. J., Osborne, J. L. & Grimm, V. Multiple stressors: using the honeybee model BEEHAVE to explore how spatial and temporal forage stress affects colony resilience. Oikos (2015).
de Miranda, J. R., Cordoni, G. & Budge, G. The Acute bee paralysis virus-Kashmir bee virus-Israeli acute paralysis virus complex. J. Invert. Path. 103, S30–S47, doi:10.1016/j.jip.2009.06.014 (2010).
Anderson, D. L. & Gibbs, A. J. Inapparent virus-infections and their interactions in pupae of the honey bee (Apis mellifera Linnaeus) in Australia. J. Gen. Virol. 69, 1617–1625 (1988).
Bailey, L. & Woods, R. D. Two more small RNA viruses from honey bees and further observations on sacbrood and acute bee paralysis viruses. J. Gen. Virol. 37, 175–182, doi:10.1099/0022-1317-37-1-175 (1977).
Anderson, D. L. Kashmir Bee Virus - a relatively harmless virus of honeybee colonies. Am. B. J. 131, 767-& (1991).
Di Prisco, G. et al. Varroa destructor is an effective vector of Israeli acute paralysis virus in the honeybee, Apis mellifera. J. Gen.Virol. 92, 151–155, doi:10.1099/vir.0.023853-0 (2011).
de Miranda, J. R. et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 52, doi:10.3896/ibra.1.52.4.22 (2013).
Tamura, K. et al. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 28, 2731–2739, doi:10.1093/molbev/msr121 (2011).
Acknowledgements
We thank the Honeybee and Pollination RD&E programs of the Rural Industries Research and Development Corporation for funding and the participation of Australian beekeepers in the study.
Author information
Authors and Affiliations
Contributions
J.M.K.R. undertook the survey fieldwork and the laboratory testing, analysed the data and wrote the first draft of the manuscript; D.L.A. conceived the study; P.A.D. designed the survey and assisted with data analysis and interpretation of results; all three authors edited and contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession Codes: All raw reads produced and used in this study were submitted to the NCBI’s Sequence Read Archive under Bioproject PRJNA357523. Assembled viral genomes described in this paper were deposited in GenBank under the accession numbers KY465671-KY465720.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roberts, J.M.K., Anderson, D.L. & Durr, P.A. Absence of deformed wing virus and Varroa destructor in Australia provides unique perspectives on honeybee viral landscapes and colony losses. Sci Rep 7, 6925 (2017). https://doi.org/10.1038/s41598-017-07290-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07290-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
