
Abstract
Spreading depolarization (SD) is a slow propagating wave of strong depolarization of neural cells, implicated in several neuropathological conditions. The breakdown of brain homeostasis promotes significant hemodynamic and metabolic alterations, which impacts on neuronal function. In this work we aimed to develop an innovative multimodal approach, encompassing metabolic, electric and hemodynamic measurements, tailored but not limited to study SD. This was based on a novel dual-biosensor based on microelectrode arrays designed to simultaneously monitor lactate and glucose fluctuations and ongoing neuronal activity with high spatial and temporal resolution. In vitro evaluation of dual lactate-glucose microbiosensor revealed an extended linear range, high sensitivity and selectivity, fast response time and low oxygen-, temperature- and pH- dependencies. In anesthetized rats, we measured with the same array a significant drop in glucose concentration matched to a rise in lactate and concurrently with pronounced changes in the spectral profile of LFP-related currents during episodes of mechanically-evoked SD. This occurred along with the stereotypical hemodynamic response of the SD wave. Overall, this multimodal approach successfully demonstrates the capability to monitor metabolic alterations and ongoing electrical activity, thus contributing to a better understanding of the metabolic changes occurring in the brain following SD.
Similar content being viewed by others


Introduction
Spreading depolarization (SD) is a slow propagating wave of massive but short-term depolarization of neuronal and glial cells, implicated in a wide spectrum of neuropathological conditions such as traumatic brain injury (TBI), subarachnoid hemorrhage, stroke, epilepsy and migraine aura1,2,3. It is triggered when a strong stimulus simultaneously depolarizes a minimum critical volume of brain tissue causing a drop in neuronal transmembrane resistance. The re-establishment of ionic gradients after SD, via activation of ATP-dependent pumps, is extremely demanding energetically. Coherently, SD is characterized by marked metabolic changes associated with increased ATP consumption accompanied by hemodynamic changes which are required to deliver metabolic substrates imposed by the increase in metabolic demand4.
In numerous neuropathological conditions, such as TBI, it is recognized that SD does not occur as an epiphenomenon, but can elicit further neuronal injury after the primary insult thus often worsening the outcome. In this regard, major relevance has been attributed to the imbalance of metabolic and vascular mechanisms required for the restoration of brain homeostasis. It is now well accepted that SD events elicit a significant decrease in extracellular glucose concentration alongside with an increase in lactate5,6,7,8,9. Importantly, the magnitude and profile of this metabolic disturbance impacts on neuronal viability and on the clinical outcome, as substantiated by the observation that persistent low glucose levels10 and increased lactate/glucose ratio11 are associated to unfavorable outcome in TBI patients. Thus, the understanding of the dynamics fluctuation of these metabolic substrates is of paramount importance for prognostication and definition of therapeutic strategies in the clinical setting12.
Our knowledge of brain metabolism has been significantly advanced by the ability to monitor neurometabolic events in vivo with high spatial, temporal and chemical resolution. Relevant information has been obtained by non-invasive neuroimaging techniques (e.g. MRI, PET and NMR) which allow us to monitor neurometabolic events without tissue disruption. While these approaches present great advantages with relation to clinical translation, they are still limited in terms of spatial and temporal resolution. Invasive approaches, such as rapid sampling microdialysis (rsMD) have also been valuable tools for monitoring metabolic changes associated with SD events both in animal models of brain injury and patients6,7,8, 13. However, the minute-to-minute time resolution limits the detection of rapid changes in the metabolic substrates. Few studies have successfully addressed neurometabolic events during SD with the higher temporal resolution (second-by-second) as that afforded by amperometric biosensors5, 8. In fact, real-time electrochemical measurements of neurochemicals in the brain extracellular space is frequently a challenging task due to the complexity of the chemical environment14.
Microelectrode arrays (MEAs) are a very attractive microelectrode platform for developing amperometric enzyme-based biosensors15. The well-defined and highly reproducible geometrical configuration of the recording sites in the MEAs is important to accomplish reproducible measurements of the targeted analytes with spatial resolution. Another advantage of MEAs is that they can be configured for multi-analyte detection16, 17. This is of major significance in cases where correlation of the dynamic changes of the analytes is intrinsically relevant, as is the case for lactate and glucose18.
In addition to the high temporal and spatial resolution, MEA-based biosensors offer another advantage as, once coupled to amperometry, they can concurrently provide local field potential (LFP)-related information. Using MEAs-based biosensors, Zhang and collaborators demonstrated that the high frequency component of the amperometric recordings is qualitatively and quantitatively correlated with LFPs under several experimental manipulations19. This approach, further validated by others20, 21, allows the correlation between neurochemical alterations and ongoing electrical activity with an unique spatial and temporal precision. The measurement of neuronal activity has been proven to be of utmost relevance to detect neurological dysfunctions in the clinical setting following TBI22. In particular, the spectral profile of the electrical events assessed by electroencephalography (EEG) and electrocortiography (EcoG) have been suggested to provide relevant information about SD, including its prediction22,23,24.
In the present study we aimed to develop an innovative multimodal approach, encompassing metabolic, electric and hemodynamic measurements, tailored to study SD events on basis of a high temporal and spatial resolution window. However, we anticipate the potential of this approach to other applications, for instance to elucidate the mechanisms underlying neurometabolic coupling. We used platinum multisite MEAs to design and develop a new dual lactate-glucose microbiosensor, which was extensively characterized to address the suitability for in vivo measurements.
We established a multimodal approach using a new ceramic MEA-based design directly implanted in the brain tissue, providing simultaneous neurometabolic and electrophysiological information. In addition, we monitored cortical cerebral blood flow by laser Doppler Flowmetry. Using this approach we successfully measured local rapid fluctuations in lactate and glucose associated with neuronal activity (LFP-related currents) in the cortex of anesthetized rats during SD.
Results
In vitro dual biosensor characterization
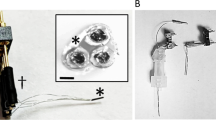
Ceramic-based MEAs (R1 configuration) with 4 in-line Pt sites were configured for simultaneous detection of glucose and lactate by individually coating two of the sites with Lactate Oxidase (LOx) and Glucose Oxidase (GOx) (Fig. 1). Figure 2A shows a representative recording of the response of the LOx-GOx microbiosensor array (referred hereinafter as LOx-GOx MBA) to successive additions of increasing concentrations of lactate and glucose. The LOx- and GOx-coated sites exhibited a significant and selective response to lactate and glucose, respectively, while no significant current changes were detected at the sentinel sites. LOx-GOx MBAs with crosstalk between sites (>2%) were discarded. The response to both substrates followed Michaelis-Menten kinetics with a range of linearity (R2 > 0.99) up to 5 and 12 mM for lactate and glucose, respectively (Fig. 2B,C). The most relevant kinetics and analytical parameters are summarized in Table 1.

Schematic representation of the dual lactate-glucose biosensor developed from ceramic-based multisite microelectrode arrays (MEA) (125 µm thick) containing 4 platinum recording sites in line (R1, 50 × 150 µm2, spacing 50 µm). Sites 1 and 3 sites (active sites) were coated with a cocktail solution containing Lactate Oxidase (LOx) or Glucose Oxidase (GOx), BSA and glutaraldehyde (GA). Sites 2 and 4 (sentinel sites) were coated with the inactive protein matrix. The sites were further modified with an exclusion layer of meta-phenylenediamine (m-PD), to improve selectivity, and a diffusional barrier of polyurethane (PU), to extend the linear range for the substrate detection.

Enzyme kinetics of LOx-PU coated sites and GOx-PU coated sites towards lactate (blue line) and glucose (orange line) detection. Grey line represents the response of the sentinel site. (A) Representative calibration showing responses of LOx-GOx MBA to successive additions of lactate and glucose. The final concentrations after each addition was 0.05, 0.1, 0.2, 0.5, 1, 2, 3, 5, 7.5, 10, 15 and 20 mM for lactate and 0.125, 0.5, 1, 2, 4, 8,12, 20, 28, 44 and 60 mM for glucose. (B,C) Calibration plot of the average steady-state current as a function of lactate (B) and glucose (C) concentration. Data were fitted to the Michaelis–Menten equation by using non-linear regression analysis. Inserts: Linear regression of the response of the LOx-PU coated sites (B) and GOx-PU coated sites (C) to lactate and glucose, respectively.
To minimize the interference of undesirable electroactive compounds, the LOx-GOx MBA sites were modified with a poly-[m-phenylenediamine] exclusion layer25. In spite of the specificity of LOx and GOx for the corresponding substrates, the presence of electroactive compounds in the brain extracellular space limits biosensor specificity, namely ascorbate which extracellular concentration range of 200–400 μM26,27,28. A typical response of the LOx-GOx MBA to lactate and glucose in the presence of high concentrations of ascorbate (0.5 mM) is depicted in Fig. 3. The response to ascorbate was negligible in active and sentinel sites when compared to the response to lactate (0.5 mM) and glucose (0.5 mM) in active sites. The selectivity ratio (substrate:ascorbate) was 52 ± 11: 1 and 98 ± 28: 1 (n = 4) and the corresponding blocking efficiency 98% and 99% for LOx-PU and GOx-PU coated sites, respectively. No measurable current changes were detected by addition of 10 µM of dopamine while all sites responded to H2O2 (10 µM), used as a test molecule. The reported selectivity can be improved by taking advantage of the “self-referencing” recording technique through the off-line subtraction of the current recorded at the sentinel sites from that of the active sites29.

Representative calibration of the LOx-GOx MBA to lactate and glucose (0.5 mM) in the presence of interferents (0.5 mM of ascorbate and 10 µM of dopamine). H2O2 was used as test substance (positive control for sentinel sites).
To further address the suitability of LOx-GOx MBA to accurately measure lactate and glucose in the brain extracellular space we evaluated the dependencies of the LOx-GOx MBA for O2, pH and temperature (Fig. 4). As expected, considering that O2 is a co-substrate for both LOx and GOx, the response to lactate and glucose (1 mM) was affected by O2 concentration. The average steady-state current response for substrates plotted as a function of O2 concentration followed a Michaelis-Menten kinetics model and the KM(O2) calculated from the fits averaged 2.5 ± 0.2 µM and 2.6 ± 0.3 µM for lactate and glucose, respectively (Fig. 4A). Furthermore, we observed that the LOx-GOx MBA response to lactate and glucose was also dependent on temperature and pH albeit that variations were relatively low and similar for both analytes: 2–3%/°C and 1–2%/pH unit from 6.5 to 7.5 (Fig. 4B,C).

Evaluation of oxygen-, temperature- and pH- dependencies of the LOx-GOx MBA. (A) Average normalized current detected by LOx-PU and GOx-PU coated sites, respectively, to 1 mM lactate and 1 mM glucose as a function of oxygen concentration. Data were fitted to the Michaelis–Menten equation by using non-linear regression analysis. (B) Effect of temperature variation on the recorded current of LOx-PU and GOx-PU coated sites in the presence of 1 mM of lactate and glucose, respectively. (C) Effect of pH on the response of LOx-PU and GOx-PU coated sites to 1 mM of lactate and glucose, respectively. The response towards glucose was fitted to a Gaussian function.
In vivo validation of lactate and glucose measurements by the dual biosensor
The capability of the LOx-GOx MBA described herein to measure lactate and glucose concentrations in the brain extracellular space was confirmed in vivo by the higher background current of the LOx and GOx-coated sites as compared to the sentinel sites (Fig. 5A). By imposing anoxic conditions, promoted by forcing the animal to breathe pure N2 gas, the amperometric currents of active sites were similar to that of the sentinel sites (note that O2 is a co-substrate for the LOx and GOx). This supports that the current recorded by LOx and GOx-coated sites results from lactate and glucose oxidation, respectively, with minimal chemical interferences.

In vivo validation of lactate and glucose measurements by the dual biosensor. (A) Recording of basal lactate (blue line) and glucose (orange line) concentrations with the LOx-GOx MBA implanted in the cerebral cortex of urethane-anesthetized rats. The detection of lactate and glucose was hampered by anoxic conditions created by forcing the animal to breath pure N2 gas (downward arrow). The induced changes in pO2 were confirmed systemically by pulse oximetry (green line). (B) In vivo recording of O2 changes (green line) and cerebral blood flow (pink line) in the cerebral cortex of urethane-anesthetized rats in response to SD. SD was induced by a needle prick at the time indicated by the downward arrow.
To define the potential interference of O2 fluctuations on the lactate and glucose measurements by LOx-GOx MBA during SD, we further measured the changes in O2 concentration in the extracellular space of the rat brain with a sentinel site polarized at a negative potential (−0.6 V vs Ag/AgCl)18. The LOx-GOx MBA was inserted in the medial parietal association cortex along with a laser Doppler probe to monitor cortical CBF. SD events were mechanically induced by a needle prick 5 mm away from the recording site as seen in Fig. 6. Continuous monitoring of physiological parameters revealed spontaneous fluctuations of heart and respiration rate and pulse oximetry within a physiological range, without correlation to SD events. The CBF was measured in close proximity of the LOx-GOx MBA to confirm the occurrence of the SD episode. Hyperemia typically starts ca. 15 s after the DC shift onset, reaching a maximum after complete repolarization4. We observed the hyperemic wave of propagating at 2–5 mm/min characterized by a robust increase in CBF (>100%) that lasted 2 min (Fig. 5B). Within the same time course, we observed a significant change in O2 associated with the hyperemic response characterized by an initial significant decrease to 25 µM and a later transient increase to 80 µM (Fig. 5B).

Schematic representation of the in vivo setup used to simultaneous measure cerebral blood flow, by laser Doppler Flowmetry, and neurometabolic and electrophysiological changes, by the LOx-GOx MBA, in response to SD depolarization induced by the needle prick of cortical surface.
Measurement of neurometabolic and electrophysiological changes during cortical spreading depolarization
The amperometric recordings with LOx-GOx MBA were performed at high frequency (100 Hz) with the aim of simultaneously measuring changes in lactate and glucose concentrations and LFP-related currents. Figure 7 shows a representative recording of CBF, the low frequency component (<1 Hz) of the amperometric signal recorded by the LOx-PU, GOx-PU and sentinel sites of the LOx-GOx MBA, and the spectral analysis of the higher frequency component (>1 Hz) of the same amperometric signal. The increase in CBF associated with the SD wave occurred along with multiphasic fluctuations in the extracellular concentration of both lactate and glucose. Lactate concentration initially decreased slightly with a minimum coincident with the time of maximal CBF. It then steeply increased to reach a maximum concentration within 4 minutes of SD induction. On average, lactate increased 66% from a basal concentration of 0.83 ± 0.22 mM (n = 3). The basal levels were restored after 20 minutes. Accompanying such changes, we observed a dramatic decline of glucose concentration that reached a minimal value within 5 minutes of SD induction. On average, glucose decreased 51% from a basal concentration of 1.56 ± 0.47 mM (n = 3). This decrease was then followed by a slower and typically incomplete recovery 20 minutes after SD induction. During the recovery period spontaneous lactate oscillations were observed, with no correlation with glucose changes. The sentinel site recorded only a negligible current change. The matched drop in glucose and rise in lactate, resulted in a transient increase of the lactate/glucose ratio of over 300% increase (from an average value of 0.7 prior to SD).

Neurometabolic changes and LFP-related information during cortical SD addressed by (A) CBF recording, (B) low frequency component (<1 Hz) of the amperometric signals recorded by the LOx-GOx MBA and (C) power spectrogram of the higher frequency component (>1 Hz) of the amperometric signal. (D) Average power density of the amperometric signals during the 60 s period recorded before (epoch a) and after the SD induction (epoch c). (E) Average relative power of delta (1–3 Hz), theta (3–6 Hz) and beta (14–30 Hz) bands in five different periods (60 s) associated with the SD event: a – control condition, b – after needle prick, c – during hyperemic response, d – after hyphemia, e – recovered condition. The events are marked in the bottom of the power spectrogram (C).
The spectral analysis of LFP-related currents retrieved from the high-frequency component of the amperometric signals revealed relevant alterations in the power spectrum in close relation with the metabolic changes elicited by SD (Fig. 7C). We observed that the SD wave was accompanied by a synchronization of the slow wave activity in delta and theta oscillations reflected by two prominent power bands, respectively, at 2 and 4 Hz (see Fig. 7C and D). The synchronization persisted for ca. 20 min and was associated with a decrease in the relative power density of that frequency range (Fig. 7E). Coincident with the hemodynamic response, we observed a significant decrease in the power density in the delta range (1–3 Hz) that lasted ca. 1 minute.
Discussion
In this work we present a multimodal approach, encompassing metabolic, electric and hemodynamic measurements, tailored to study spreading depolarization events. For that purpose we developed, characterized and validated a dual microbiosensor array for lactate and glucose measurements in rat brain tissue with high temporal, spatial and chemical resolution. The detailed in vitro characterization revealed that the dual glucose and lactate oxidase- microbiosensor array, based on an efficient glutaraldehyde immobilization procedure following polyurethane coating, exhibit suitable analytical performance towards measurements in brain tissue, in particular a good linear range, high sensitivity and selectivity. In particular the sensitivity and linear range are comparable with data previously reported for planar microbiosensors30, 31 and Pt-wire-based microbionsensors32 similarly modified with phenylenediamines and polyurethane. The polyurethane membrane is required to extend the linear range, which is critical for lactate measurement considering the low apparent KM of LOx33. Coherently, polyurethane promoted a significant increase in KM both for lactate (from 1.4 ± 0.2 mM to 22 ± 6 mM, unpublished) and glucose (from 6.6 ± 0.2 mM29 to 80 ± 14 mM), in accordance to data reported by others16, 30, 32. This effect is consequence of the diffusion limitation of analyte versus O2 imposed by the polyurethane layer and unlikely due to an actual change in enzyme kinetics. Overall, the LOx-GOx-MBA array exhibited an appropriate dynamic range to measure large changes in lactate and glucose concentration in brain extracellular space. This expanded linear range was obtained at the expense of a decrease in sensitivity and a slower response time as expected due to the diffusion barrier imposed by the polyurethane membrane, yet it is in a range suitable for the detection of neurometabolic events on a second-by-second time-scale. A much lower dynamic range was reported for Pt/Ir wire-based biosensors used for the same purpose, which indeed is unarticulatable with the magnitude of the glucose and lactate changes reported5. The use of polyurethane offers an additional advantage related to a reduction of oxygen dependency obtained by the reduction in the diffusion of the metabolic substrates to the enzymatic matrix relative to that of O2 15, 31. This is of major relevance considering the evidence for relevant changes in pO2 accompanying SD events34. Coherently, LOx-GOx-MBA array revealed minimal oxygen-dependencies in lactate and glucose measurements. Data support that while increases above physiological concentrations of O2 (30–50 µM)35, 36 unlikely influence lactate and glucose measurements, decreasing O2 below 5 µM could interfere with the amperometric current measurements. The amplitude of O2 changes observed in vivo in response to the same experimental paradigm used to evaluate the metabolic and electrophysiological changes during SD, supports that the changes in O2 concentrations in the brain extracellular space are unlikely to impact lactate and glucose measurements by the LOx-GOx MBA during SD. This further supports the suitability of this array to monitor metabolic changes during SD. Furthermore, lactate and glucose measurements by LOx-GOx-MBA array also revealed minimal dependency of temperature and pH. This is of major relevance, as while these experimental parameters are kept constant in in vitro studies, significant changes can occur in vivo with impact on the estimation of lactate and glucose concentrations. For instance, there is evidence for pH decreases (~0.4 pH unit)9 and temperature oscillations37 during SD. Importantly, the magnitude of changes induced by this variables (within a reasonable window of variation) was minimal and in the same extension for both lactate and glucose, thus unlikely to significantly impact on our measurements. In situations expected to cause more dramatic changes in O2, pH and temperature, the magnitude of the changes in these parameters should be determined and used to correct the measurements of lactate and glucose for those conditions. In this context, we must caveat that our experimental paradigm of SD was associated with a hemodynamic response mainly characterized by a dominant hyperemic response, which is typically observed in healthy and well perfused brain tissue. In injured brains, depending on the type of injury and its severity, SD events may be instead associated to an inverse neurovascular coupling38,39,40.
By using amperometric recordings in vivo with the same LOx-GOx-MBA array we successfully measured the metabolic signature of SD simultaneously with pronounced changes in the spectral profile of LFP-related currents, in correlation with CBF changes induced by a needle prick model of traumatic brain injury. In spite of some differences in the magnitude of the changes and the temporal profile, the variations in lactate and glucose here reported followed the known metabolic signature of SD, globally characterized by a fall in glucose extracellular concentration and an increase in lactate5,6,7,8,9. The significant drop in glucose concentration occurs due to glycolysis stimulation in response to a massive increase in oxidative phosphorylation to generate the ATP required to restore the ionic gradients lost during the SD wave. In spite of the increase in glucose supply by the hyperemic response, the energetic demands are such that this input is insufficient to meet demand. The accumulation of lactate in the brain extracellular space, while accepted, is more puzzling to understand, particularly under conditions of available O2 41 . The observed relative changes in lactate are in agreement with those reported using rsMD6,7,8, but the higher temporal resolution of the LOx-GOx-MBA array revealed multiphasic and more complex fluctuations in lactate extracellular concentration. In spite of different amplitudes, the biphasic change in lactate associated with SD was previously detected using electrochemical biosensors directly implanted in the brain parenchyma5. In turn, the magnitude of the changes here reported for glucose were higher than those revealed by rsMD6,7,8. The larger changes in metabolites detected using biosensors are suggested to occur due to a low-pass filtering of the fast neurometabolic changes in rsMD5. However, it should be stressed that these comparisons are of relative value considering the experimental differences, both the method for metabolites monitoring, but also in the anesthetic used, the stimuli for SD induction and the strain and age of the rats studied. The matched drop in glucose and rise in lactate, resulted in a transient increase of the lactate/glucose ratio which is suggested to be a biomarker of poor outcome in TBI and other neuropathological conditions11, 42, 43.
A major observation in this work was the marked alterations in the spectral profile of LFP-related currents accompanying the metabolic changes during and after SD. Most significantly, we observed a synchronization of the slow wave activity in the delta and theta range accompanying the period of metabolic disturbance. This time window is within the range of the period of the depression of spontaneous and evoked synaptic activity following SD4. Interestingly, alterations in delta and theta activity were also observed in the EEG of patients during attacks of migraine with prolonged aura44, a condition associated to SD events45. Furthermore, we observed a significant decrease in the power of the delta band during hemodynamic response. We speculate that this decrease is a fingerprint of the DC negative shift and may be used as surrogate marker for the SD wave. In agreement, it was shown that >80% of SDs recorded from EcoG in brain trauma manifest as depressions of high-amplitude delta activity in EEG recording24. This evidence supports the possibility of identifying SD waves by spectral analysis without recording DC potential as suggested by others46. Yet, simultaneous measurements of DC and amperometric recordings required to validate this hypothesis. In the recent years, emerging evidence has been focused on the changes in EEG profile induced by SD, particularly in the context of TBI, but a clear picture is still to be defined22.
While our observations focused on the response to a first SD triggered event, we do not exclude the occurrence of prior SD events considering that craniotomy and microelectrode insertion are reported to potentially cause SD47, 48. Indeed, a monophasic CBF increase without detectable initial hypoperfusion or relevant post-oligemia similar to that reported here is usually considered a fingerprint of subsequent SD events4. This is relevant considering that in addition to hemodynamic response, the metabolic changes and the spectral profile may vary with the number of SD events13, 49.
Overall, in this work we successfully demonstrated the innovative capability to monitor metabolic alterations and ongoing network electrical activity with a unique spatial and temporal resolution using a single array. This multimodal approach is a valuable tool with the potential to be used in basic research to expand the understanding of SD events and their impact on neuronal function and, in the future, to be adapted for clinical setting translation assisting in prognosis and definition of personalized therapeutic strategies40. Finally, we anticipate the application of this multimodal approach to not be limited to the SD context. Indeed, in the brain, this approach, coupled to pharmacology, can be useful to provide relevant insights that help to deep the knowledge of the neurovascular and neurometabolic coupling both in physiological and in pathological conditions30, 38. Another potential application is cancer, in which LOx-GOx-MBA can pave the way to novel experimental approaches to evaluate metabolic profiling in tumor microenvironment32.
Methods
Chemicals and solutions
Lactate Oxidase (LOx) (EC 1.13.12.4) from Pediococcus sp in powder form, Glucose Oxidase (GOx) (EC 1.1.3.4, Type VII) from Aspergillus niger in powder form, meta-phenylenediamine (m-PD), polyurethane, tetrahydrofuran, sodium lactate, D-(+)-Glucose, ascorbic acid, dopamine, hydrogen peroxide, bovine serum albumin (BSA), glutaraldehyde solution (25%) and urethane were obtained from Sigma-Aldrich. All other reagents were purchased from Merck, unless otherwise specified. All solutions were prepared in bi-deionized MilliQ water with resistivity ≥18 MΩ cm (Millipore Corporation, USA). The electrolyte for in vitro analytical evaluation of microbiosensors was phosphate buffer saline (PBS) 0.05 M (pH 7.4) with the following composition (mM): 100 NaCl, 10 NaH2PO4 and 40 Na2HPO4. For the pH dependency studies the pH of PBS was priorly adjusted from 5.5 to 8.5 using HCl or NaOH solutions. Stock solutions of glucose (1 M) were allowed to equilibrate the β/α anomeric ratio for 24 h before use.
Preparation and modification of Lactate-Glucose microbiosensor arrays
Ceramic-based MEAs were obtained from Center for Microelectrode Technology, University of Kentucky, USA. The R1 design configuration containing 4 sites (50 × 150 µm2) in-line separated by 50 µm was used as the microelectrode platform for construction of the dual microbiosensor array (Fig. 1). Lactate (LOx) and Glucose oxidase (GOx) were immobilized differentially onto the Pt sites using a coating procedure essentially as described29, 31. The cocktail solutions of enzymes were differentially applied to the sites surface using a microsyringe mounted on micromanipulator under a stereomicroscope using the following scheme: site 1 - LOx (3 mg/mL), BSA (1%) and glutaraldehyde (0.125%) in water and site 3 – GOx (1 mg/mL), BSA (1%) and glutaraldehyde (0.125%) in water. The remaining sites (2 and 4) were coated with the inactive protein matrix solution containing BSA (1%) and glutaraldehyde (0.125%) using the same procedure (Fig. 1). These recording sites should be insensitive to both lactate and glucose and as such act as sentinel or null sites. The dual microbiosensors were stored dry in the dark and protected from dust, at room temperature, for at least three days to allow for curing and stabilization of the active and non-active matrices. Then, a polyurethane membrane was deposited by dip coating in a solution of polyurethane 2% dissolved in a mixture of tetrahydrofluran (98%) and dimethylformamide (2%) and allowed to dry for 24 hours. To minimize access of undesirable electrochemically active compounds to the Pt recording sites, the MEA-based GOx biosensors were modified, before use, with an exclusion layer of m-PD. Briefly, the m-PD solution (10 mM) was freshly prepared in deoxygenated PBS and electropolymerized at the MEA Pt surface using the FAST16mkIII high-speed electrochemical system (Quanteon, Nicholasville, KY, USA) by cyclic voltammetry between +0.25 and +0.75 V vs an Ag/AgCl reference electrode (RE-5, BAS Inc., USA) at a scan rate of 50 mV/s during 20 minutes. For some in vitro studies, S2 MEA design configuration was used as a microelectrode platform for biosensor construction, without the use of sentinel sites.
In vitro evaluation and characterization
The in vitro evaluation of the LOx-GOx MBA for measurement of lactate and glucose was performed by amperometry at +0.7 V vs Ag/AgCl (RE-5, BAS Inc., USA) using the FAST16mkIII high-speed electrochemical system in a two-electrode configuration mode. The experiments were carried out in 40 mL PBS 0.05 M at 37 °C under gentle stirring after a 30 minute period of current stabilization. Analytical and kinetic parameters were determined after baseline stabilization by adding aliquots of the stock lactate or glucose solution to obtain final concentrations in the range of 0.05 to 20 mM and 0.125 to 60 mM, respectively. The sensitivity to lactate and glucose, selectivity against major interferents and the sensitivity to the reporter molecule were determined by three additions of lactate and glucose (0.5 mM) in the presence of ascorbic acid 0.5 mM, followed by DA 10 µM and H2O2 10 µM.
To evaluate the dependency of LOx-GOx MBAs for O2, temperature and pH, the responses to glucose and lactate (1 mM) were measured under variable O2 concentrations, temperature and pH, essentially as previously described29.
Surgical procedures and in vivo experiments
All the procedures used in this study were performed in accordance with the European Union Council Directive for the Care and Use of Laboratory animals, 2010/63/EU and were approved by the local ethics committee of the animal house facilities of the Center for Neurosciences and Cell Biology (ORBEA - Órgão Responsável pelo Bem-Estar Animal). In vivo studies were carried out on adult male Wistar rats (10 weeks) maintained in controlled environmental conditions: temperature of 22–24 °C, relative humidity of 45–65%, 15 air exchanges per hour and a 12:12 light/dark cycle. Animals were housed in filter-topped type III Makrolon cages on an individually ventilated caging system (VentiRack Bioscreen™). Rats were fed with a standard rat chow diet (4RF21-GLP Mucedola, SRL, Settimo Milanese, Italy) and were provided with chlorinated water, both ad libitum.
Rats were anesthetized with urethane (1.25 g/kg, i.p.) and a stereotaxic surgery was performed as previously described29. Animals breathed spontaneously and their body temperature was maintained at 37 °C using a heated pad coupled to a Gaymar Heating Pump (Braintree Scientific, Inc., USA). Basic physiological parameters (blood O2 saturation, heart rate and breath rate) were continuously monitored during the experiment using a pulse oximeter system (MouseOx®, Starr life sciences, Oakmont, PA, USA). After exposing the skull, two craniotomies were performed: a primary posterior craniotomy overlying the parietal cortex (2.5 to 4.5 mm posterior and 1.5 to 3.5 mm lateral to bregma) for measurements and a secondary craniotomy (~1 mm2, 5 mm posterior to the primary craniotomy) for SD induction. The meninges were removed from the brain surface prior to the insertion of the pre-calibrated LOx-GOx MBA into the rat cerebral cortex according to coordinates calculated from bregma based on the rat brain atlas of Paxinos and Watson50: 3.6 mm posterior, 2.0 mm lateral and 1.8 mm ventral to bregma. The LOx-GOx MBA was positioned with the site’s surface in posterior orientation. An Ag/AgCl reference electrode prepared from a silver wire (200 µm diameter) was placed underneath the retracted skin and kept moistened with saline. Electrochemical recordings were performed using the FAST16mkIII high-speed electrochemical system (Quanteon, Nicholasville, KY, USA) applying a constant potential (+0.7 V vs Ag/AgCl) and using a 100 Hz acquisition rate. Cerebral blood flow was simultaneous and continuously measured by laser Doppler flowmetry (Periflux System 5000, Perimed AB, Järfälla, Sweden). The LDF probe (780 nm wavelength, 250 µm fiber separation) was placed on the cortical surface in the immediacy of the MBA. Calibration of the probe was performed routinely according to manufacturer recommendations to equalize the perfusion values among the various recordings. The time constant was set to 0.03 s and the signal-processing unit used a bandwidth of 32 Hz. After a 1 hour stabilization period, SD was induced 5 mm away from the recording site by pricking the upper cortical layers with the tip of a 26-gauge needle as previously described6, 8. As a control experiment, anoxia was induced by forcing the animal to breath pure N2 gas until cardiac arrest was achieved.
Data Analysis
In vitro data analysis was performed using GraphPad Prism 5.0. For the kinetic analyses, data were fitted to a Michaelis-Menten type equation and the maximum steady-state current response normalized by the geometric area of the active Pt site (Jmax) and apparent Michaelis-Menten constants (KM) for lactate and glucose were determined. The selectivity ratio, on a molar basis, against ascorbate was calculated from the recording of the enzyme-coated sites as the ratio of the sensitivities for lactate or glucose and ascorbate. The crosstalk between sites is defined by the in vitro response detected in an inactive site (sentinels) following the addition of glucose or lactate The limit of detection (LOD) was defined as the concentration that corresponds to a signal-to-noise ratio of 3. The response time (t90%
- t10%) of the microbiosensors was determined by fitting the data to a Boltzmann sigmoid function and calculating the time interval between 10% and 90% of the maximal response to lactate or glucose (1 mM). For the O2 dependency studies, the apparent Michaelis-Menten constant for O2 (KM(O2)) was determined under fixed lactate and glucose concentrations by fitting the normalized data to a Michaelis-Menten type equation. Values are given as the mean ± standard error of the mean (SEM).
The signal processing of in vivo recordings was performed using OriginPro2016. The amperometric recordings were low passed filtered at 1 Hz to extract the electrochemical component of the oxidation currents associated to lactate, glucose and potential interferents. Power spectrograms were constructed using the short-time Fourier transform of the unprocessed amperometric signals. The power density relative to the periods before and after SD was calculated by averaging the Fast Fourier transform of 60 s epochs. The relative powers of delta (1–3 Hz), theta (3–6 Hz) and beta (14–30 Hz) bands were calculated by ratio of the power of the band and total power.
References
Lauritzen, M. et al. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 31, 17–35, doi:10.1038/jcbfm.2010.191 (2011).
Kramer, D. R., Fujii, T., Ohiorhenuan, I. & Liu, C. Y. Cortical spreading depolarization: Pathophysiology, implications, and future directions. J Clin Neurosci 24, 22–27, doi:10.1016/j.jocn.2015.08.004 (2016).
Dreier, J. P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17, 439–447, doi:10.1038/nm.2333 (2011).
Ayata, C. & Lauritzen, M. S. Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol Rev 95, 953–993, doi:10.1152/physrev.00027.2014 (2015).
Balanca, B. et al. Altered hypermetabolic response to cortical spreading depolarizations after traumatic brain injury in rats. J Cereb Blood Flow Metab. doi:10.1177/0271678X16657571 (2016).
Feuerstein, D. et al. Regulation of cerebral metabolism during cortical spreading depression. J Cereb Blood Flow Metab. doi:10.1177/0271678X15612779 (2015).
Hashemi, P. et al. Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leao’s spreading depression. J Cereb Blood Flow Metab 29, 166–175, doi:10.1038/jcbfm.2008.108 (2009).
Rogers, M. L. et al. Continuous online microdialysis using microfluidic sensors: dynamic neurometabolic changes during spreading depolarization. ACS Chem Neurosci 4, 799–807, doi:10.1021/cn400047x (2013).
Scheller, D., Kolb, J. & Tegtmeier, F. Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression. Neurosci Lett 135, 83–86 (1992).
Vespa, P. M. et al. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: a microdialysis study. J Cereb Blood Flow Metab 23, 865–877, doi:10.1097/01.WCB.0000076701.45782.EF (2003).
Timofeev, I. et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain: a journal of neurology 134, 484–494, doi:10.1093/brain/awq353 (2011).
Dreier, J. P. et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J Cereb Blood Flow Metab. doi:10.1177/0271678X16654496 (2016).
Parkin, M. et al. Dynamic changes in brain glucose and lactate in pericontusional areas of the human cerebral cortex, monitored with rapid sampling on-line microdialysis: relationship with depolarisation-like events. J Cereb Blood Flow Metab 25, 402–413, doi:10.1038/sj.jcbfm.9600051 (2005).
Parkin, M. C., Hopwood, S. E. & Boutelle, M. G. Resolving dynamic changes in brain metabolism using biosensors and on-line microdialysis. Trends in Analytical Chemistry 22, 487–497 (2003).
Weltin, A., Kieninger, J. & Urban, G. A. Microfabricated, amperometric, enzyme-based biosensors for in vivo applications. Anal Bioanal Chem 408, 4503–4521, doi:10.1007/s00216-016-9420-4 (2016).
Burmeister, J. J. et al. Ceramic-based multisite microelectrode arrays for simultaneous measures of choline and acetylcholine in CNS. Biosens Bioelectron 23, 1382–1389, doi:10.1016/j.bios.2007.12.013 (2008).
Vasylieva, N., Marinesco, S., Barbier, D. & Sabac, A. Silicon/SU8 multi-electrode micro-needle for in vivo neurochemical monitoring. Biosens Bioelectron 72, 148–155, doi:10.1016/j.bios.2015.05.004 (2015).
Ledo, A. et al. Ceramic-Based Multisite Platinum Microelectrode Arrays: Morphological Characteristics and Electrochemical Performance for Extracellular Oxygen Measurements in Brain Tissue. Analytical chemistry 89, 1674–1683, doi:10.1021/acs.analchem.6b03772 (2017).
Zhang, H., Lin, S. C. & Nicolelis, M. A. Acquiring local field potential information from amperometric neurochemical recordings. J Neurosci Methods 179, 191–200, doi:10.1016/j.jneumeth.2009.01.023 (2009).
Disney, A. A., McKinney, C., Grissom, L., Lu, X. & Reynolds, J. H. A multi-site array for combined local electrochemistry and electrophysiology in the non-human primate brain. J Neurosci Methods 255, 29–37, doi:10.1016/j.jneumeth.2015.07.009 (2015).
Viggiano, A., Marinesco, S., Pain, F., Meiller, A. & Gurden, H. Reconstruction of field excitatory post-synaptic potentials in the dentate gyrus from amperometric biosensor signals. J Neurosci Methods 206, 1–6, doi:10.1016/j.jneumeth.2012.01.013 (2012).
Rapp, P. E. et al. Traumatic brain injury detection using electrophysiological methods. Front Hum Neurosci 9, 11, doi:10.3389/fnhum.2015.00011 (2015).
Hertle, D. N. et al. Changes in electrocorticographic beta frequency components precede spreading depolarization in patients with acute brain injury. Clin Neurophysiol 127, 2661–2667, doi:10.1016/j.clinph.2016.04.026 (2016).
Hartings, J. A. et al. Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol 76, 681–694, doi:10.1002/ana.24256 (2014).
Murphy, L. M. Reduction of Interference Response at a Hydrogen Peroxide Detecting Electrode Using Electropolymerized Films of Substituted Naphthalenes. Anal Chem 70, 2928–2935, doi:10.1021/ac971182r (1998).
Stamford, J. A., Kruk, Z. L. & Millar, J. Regional differences in extracellular ascorbic acid levels in the rat brain determined by high speed cyclic voltammetry. Brain research 299, 289–295 (1984).
Miele, M. & Fillenz, M. In vivo determination of extracellular brain ascorbate. J Neurosci Methods 70, 15–19 (1996).
Ferreira, N. R., Lourenço, C. F., Barbosa, R. M. & Laranjinha, J. Real time in vivo measurement of ascorbate in the brain using carbon nanotube-modified microelectrodes. Electroanalysis 25, 1757–1763 (2013).
Lourenço, C. F., Ledo, A., Laranjinha, J., Gerhardt, G. A. & Barbosa, R. M. Microelectrode array biosensor for high-resolution measurements of extracellular glucose in the brain. Sensors and Actuators B: Chemical 237, 298–307, doi:10.1016/j.snb.2016.06.083 (2016).
Weltin, A., Enderle, B., Kieninger, J. & Urban, G. A. Multiparametric, Flexible Microsensor Platform for Metabolic Monitoring In Vivo. Ieee Sens J 14, 3345–3351, doi:10.1109/Jsen.2014.2323220 (2014).
Burmeister, J. J., Palmer, M. & Gerhardt, G. A. L-lactate measures in brain tissue with ceramic-based multisite microelectrodes. Biosens Bioelectron 20, 1772–1779, doi:10.1016/j.bios.2004.07.003 (2005).
Rocchitta, G. et al. Simultaneous telemetric monitoring of brain glucose and lactate and motion in freely moving rats. Analytical chemistry 85, 10282–10288, doi:10.1021/ac402071w (2013).
Taurino, I. et al. Comparative study of three lactate oxidases from Aerococcus viridans for biosensing applications. Electrochimica Acta 93, 72–79, doi:10.1016/j.electacta.2013.01.080 (2013).
Piilgaard, H. & Lauritzen, M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 29, 1517–1527, doi:10.1038/jcbfm.2009.73 (2009).
Nair, P. K., Buerk, D. G. & Halsey, J. H. Jr. Comparisons of oxygen metabolism and tissue PO2 in cortex and hippocampus of gerbil brain. Stroke 18, 616–622 (1987).
Murr, R., Berger, S., Schurer, L., Peter, K. & Baethmann, A. A novel, remote-controlled suspension device for brain tissue PO2 measurements with multiwire surface electrodes. Pflugers Arch 426, 348–350 (1994).
LaManna, J. C., McCracken, K. A., Patil, M. & Prohaska, O. J. Stimulus-activated changes in brain tissue temperature in the anesthetized rat. Metab Brain Dis 4, 225–237 (1989).
Hinzman, J. M. et al. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain: a journal of neurology 137, 2960–2972, doi:10.1093/brain/awu241 (2014).
Ostergaard, L. et al. Neurovascular coupling during cortical spreading depolarization and -depression. Stroke 46, 1392–1401, doi:10.1161/STROKEAHA.114.008077 (2015).
Woitzik, J. et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 80, 1095–1102, doi:10.1212/WNL.0b013e3182886932 (2013).
Dienel, G. A. Lactate shuttling and lactate use as fuel after traumatic brain injury: metabolic considerations. J Cereb Blood Flow Metab 34, 1736–1748, doi:10.1038/jcbfm.2014.153 (2014).
Cornford, E. M. et al. Regional analyses of CNS microdialysate glucose and lactate in seizure patients. Epilepsia 43, 1360–1371 (2002).
Goodman, J. C., Valadka, A. B., Gopinath, S. P., Uzura, M. & Robertson, C. S. Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit Care Med 27, 1965–1973 (1999).
Ogunyemi, A. O. Migraine with prolonged aura: correlation of clinical and EEG features. Behav Neurol 8, 109–114, doi:10.3233/BEN-1995-8206 (1995).
Lauritzen, M. Pathophysiology of the migraine aura. The spreading depression theory. Brain: a journal of neurology 117((Pt 1)), 199–210 (1994).
Koroleva, V. I., Davydov, V. I. & Roshchina, G. Y. Suppression of EEG gamma activity–an informative measure of spreading depression waves in the neocortex of the conscious rabbit. Neurosci Behav Physiol 36, 625–630, doi:10.1007/s11055-006-0067-z (2006).
Chang, J. C. et al. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain: a journal of neurology 133, 996–1012, doi:10.1093/brain/awp338 (2010).
Tomida, S., Wagner, H. G., Klatzo, I. & Nowak, T. S. Jr. Effect of acute electrode placement on regional CBF in the gerbil: a comparison of blood flow measured by hydrogen clearance, [3H]nicotine, and [14C]iodoantipyrine techniques. J Cereb Blood Flow Metab 9, 79–86, doi:10.1038/jcbfm.1989.11 (1989).
Roshchina, G. Y., Koroleva, V. I. & Davydov, V. I. Effects of the Functional State of the Brain Evoked by Passage of a Spreading Depression Wave on the Properties of Subsequent Wave. Neurosci Behav Physiol 44, 134–140, doi:10.1007/s11055-014-9887-4 (2014).
Paxinos, G. & Watson, C. The Rat Brain in Sterotaxic Coordinates. Academic Press, San Diego (2007).
Acknowledgements
This work was funded by FEDER funds through the Operational Program Competitiveness Factors - COMPETE and national funds by FCT - Foundation for Science and Technology under strategic project POCI-01-0145-FEDER-007440. C.F.L. acknowledges fellowship SFRH/BPD/82436/2011 from FCT. We acknowledge some support from Quanteon, LLC for salary of A.L.
Author information
Authors and Affiliations
Contributions
C.F.L. conceived the study, conducted the experiments, analyzed the data and wrote the manuscript, A.L. contributed to study design, assisted experiments and data analysis, and revised the manuscript, G.A.G. and J.L. discussed and revised the manuscript, and R.M.B. conceived and supervised the study, wrote, discussed and revised the manuscript. All authors approved the final of the version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
G.A.G. is the sole proprietor of Quanteon, LLC which manufactures the Fast16 recording system used for control of the MEA technology.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lourenço, C.F., Ledo, A., Gerhardt, G.A. et al. Neurometabolic and electrophysiological changes during cortical spreading depolarization: multimodal approach based on a lactate-glucose dual microbiosensor arrays. Sci Rep 7, 6764 (2017). https://doi.org/10.1038/s41598-017-07119-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07119-6
This article is cited by
-
Amyloid-β42 stimulated hippocampal lactate release is coupled to glutamate uptake
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
