
Abstract
Chagas disease results from infection by Trypanosoma cruzi and is a neglected tropical disease (NTD). Although some treatment drugs are available, their use is associated with severe problems, including adverse effects and limited effectiveness during the chronic disease phase. To develop a novel anti-Chagas drug, we virtually screened 4.8 million small molecules against spermidine synthase (SpdSyn) as the target protein using our super computer “TSUBAME2.5” and conducted in vitro enzyme assays to determine the half-maximal inhibitory concentration values. We identified four hit compounds that inhibit T. cruzi SpdSyn (TcSpdSyn) by in silico and in vitro screening. We also determined the TcSpdSyn–hit compound complex structure using X-ray crystallography, which shows that the hit compound binds to the putrescine-binding site and interacts with Asp171 through a salt bridge.
Similar content being viewed by others
Introduction
Chagas disease results from infection by Trypanosoma cruzi and is a neglected tropical disease (NTD)1. This disease is endemic to approximately 20 countries, including the southern United States and parts of Latin America1,2,3, and the World Health Organization (WHO) estimates that as many as 10–13 million people are chronically infected with this disease3, 4. Moreover, approximately 90 million people are exposed to the risk of infection annually, and 21,000 deaths are reported each year3,4,5. Accordingly, Chagas disease remains a serious vector-borne infection6.
The infection routes of T. cruzi primarily occur through contact with the feces of blood-sucking Triatominae and blood transfusion7. Chagas disease is classified into acute and chronic phases8, 9. The acute phase is characterized by high blood parasitemia and fever. In some cases, myocarditis or meningoencephalitis is caused by parasitization in the cardiac muscle or the brain during acute infection. The chronic phase results from an untreated acute phase, and approximately 20–30% of chronical infections are symptomatic3, 4, 8.
Currently, two drugs developed during the 1960s—nifurtimox and benznidazole—are used to combat T. cruzi infections. These drugs are effective during both the acute and early chronic phases. However, their use is associated with serious side effects, and they exhibit limited efficacy during the chronic phase of Chagas disease7, 8, 10. Therefore, the development of new anti-Chagas drugs is required.
To this end, we focused on spermidine synthase (SpdSyn) as the target protein, as sourced from the iNTRODB system11. Detailed information regarding the search method is presented in Supplementary Figure 1. The in-house web-system iNTRODB facilitates the selection of drug target proteins for NTDs, particularly for trypanosomiasis. This system provides information on trypanosomal proteins with useful annotations, including the protein structure from the Protein Data Bank (PDB) and the protein inhibitors from ChEMBL. Using this system, a user can rank the records by specific features and, thereby, identify possible candidates. When selecting SpdSyn as the target, we focused on features such as RNA interference target sequencing (RIT-seq) screening results and protein crystal structure availability. The RIT-seq screening results are taken from TriTrypDB12, which is integrated into iNTRODB. The screen shows that SpdSyn is important for survival in a related species (i.e., Trypanosoma brucei). Additionally, a structural examination of the protein by X-ray crystallography, which is useful in structure-based drug design (SBDD), is feasible since several successful structure determinations have been reported13. These two properties suggest that the inhibition of SpdSyn is a probable mechanism of action (MOA) for the treatment of Chagas disease and that the SBDD approach for inhibitor design is viable.
The polyamine biosynthesis pathway is an attractive target for the development of new antiparasitic agents. Polyamines adopt various roles in trypanothione biosynthesis and stabilization and replication of the DNA structure14,15,16,17. Genetic experimental data have shown that SpdSyn is essential for maintaining the viability of a parasite during T. brucei gene knockout or RNA interference (RNAi) studies18,19,20,21,22,23. This protein is classified as an enzyme that participates in the polyamine synthesis pathway using decarboxylated S-adenosylmethionine (dcSAM) as an aminopropyl donor. The enzyme catalyzes the transfer of polyamine from dcSAM to putrescine giving spermidine24,25,26. Importantly, the SpdSyn gene is conserved among protozoan parasites, such as T. cruzi and Leishmania spp. (TcSpdSyn: PDB ID code: 3BWB and 3BWC, Leishmania major SpdSyn: UniProt ID Q95Z84). Supplementary Figure 2 shows good alignment of a multiple amino acid sequences of trypanosomatid SpdSyns, with high identity, when compared to human SpdSyn, leads to a lower identity (67%, 60%). Although the polyamine biosynthesis pathway has been identified as a potential drug target pathway in T. brucei, Leishmania spp., and T. cruzi, the druggability of SpdSyn in T. cruzi remains inconclusive. To determine whether TcSpdSyn is a druggable enzyme, a specific inhibitor is required to test the effect of polyamine biosynthesis inhibition on the parasite using small molecules.
The crystal structure of the complex of TcSpdSyn and a substrate analogue, trans-4-methylcyclohexylamine (4MCHA), has been determined27. Figure 1 shows the binding mode of 4MCHA in the crystal structure (PDB ID code: 4YUW). This ligand interacts with Asp171 through a salt bridge and has been demonstrated to inhibit TcSpdSyn with a half-maximal inhibitory concentration (IC50) of 1.7 μM27. Asp171 is a part of the gatekeeping loop which is important in substrate binding28. Hence, SpdSyn inhibitors could interact with Asp171 to bind in the SpdSyn active site. However, because 4MCHA is too small in comparison with other pharmacologically active compounds and has only one hydrogen bond donor, this ligand would have low specificity, and its optimization as an anti-Chagas drug would be difficult. So, we decided to take a different approach to develop an anti-Chagas drug with optimal properties.

The crystal structure of the TcSpdSyn putrescine-binding site with trans-4-methylcyclohexylamine (4MCHA) (PDB ID code: 4YUW). 4MCHA binds to the putrescine-binding site and interacts with Asp171 through a salt bridge. A water molecule interacts with the 4MCHA amino group.
Docking simulation is a representative SBDD technique and is a computational method used to search for drug candidates29, 30. This simulation evaluates how well a compound fits into a binding pocket of the target protein by calculating a score function (e.g., the strength of the interactions between the compound and the target protein). Docking simulations can be complemented with data obtained through wet experimental methods to produce a compound hit rate31, which is a powerful tool for identifying new inhibitors with different core structures13.
The fragment molecular orbital (FMO) method32 conducts ab initio quantum mechanical calculations on large biomolecules, such as protein–ligand complexes, and is not only useful in identifying important interacting residues but also in determining the binding energy between a ligand and a target protein32. This method has proven to be useful in the design of new drugs33,34,35,36,37,38,39.
In the present study, we performed docking simulations to identify compounds that inhibit TcSpdSyn through binding to the putrescine-binding site. We evaluated the inhibition activity of the compounds selected from the docking simulations using an in vitro assay and performed X-ray co-crystallography to determine the TcSpdSyn structure in complex with the hit compounds. Furthermore, we conducted FMO interaction analysis to determine important interactions between TcSpdSyn and the inhibitors.
Results
In silico screening
To find potential TcSpdSyn inhibitors, we conducted a docking simulation using the TcSpdSyn X-ray structure (PDB ID code: 3BWC). The target site in the docking simulation was set to the putrescine-binding site where the catalytic reaction occurs. The compounds used for the docking simulation were sourced from a database of commercially available compounds. Figure 2 shows the docking poses of the top five potential inhibitors, which had docking scores of −8.36, −7.78, −7.72, −7.71, and −7.63. The docking results varied on the basis of target cavity size and binding compound volume. Therefore, the molecular weights and logP values for 1,000 compounds were lower than the average values of the 4,800,000 drug-like compounds used for docking simulations.

Results of the TcSpdSyn-ligand docking analysis. Docking poses of the top five compounds (green: docking results, pink: dcSAM). This simulation was conducted with the TcSpdSyn X-ray structure (PDB ID code: 3BWC). The docking results show that all compounds bind to the putrescine-binding site and that dcSAM binding in its own site.
In vitro assay and structure determination
To identify TcSpdSyn inhibitors, we performed an in vitro enzyme inhibition assay on 176 compounds sourced from the chemical compound library at Astellas Pharma. The inhibition of TcSpdSyn by the test compounds was measured at a final concentration range between 84.5 and 500 μM, depending on compound availability and solubility. Subsequently, the IC50 values of the compounds that showed >40% inhibition were determined. Several compounds with dose-response activity were found, and here, we report four compounds with IC50 values between 28 and 124 μM. Table 1 shows the parent chemical structures of active compounds and their IC50 values (the fitting curve for the IC50 calculation is shown in Supplementary Figure 3). The docking scores of the hit compounds fall within the top 2,000 compounds out of 4,800,000 drug-like compounds. Compounds 1, 3, and 4 contain an amino-alkyl chain connected to an aromatic ring, as is found in putrescine. Therefore, these amino groups should interact with Asp171 through a salt bridge.
X-ray crystal structures of TcSpdSyn-inhibitor complexes were used to confirm the binding modes of the hit compounds. Although all active compounds were subjected to X-ray crystallography, only the structure of the TcSpdSyn complex with compound 1 was determined. Co-crystallization with the remaining compounds were unsuccessful after repeated attempts, possibly due to alternate binding at a different location, such as the dcSAM binding site.
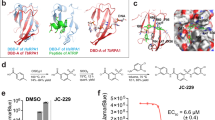
Figure 3A and C and Table 2 show the crystal structure of TcSpdSyn in complex with Compound 1 (IC50 = 124 μM). This data confirms that Compound 1 is bound to the putrescine-binding site. Figure 3B shows that compound 1 is proximally bound to Tyr73, Tyr237, Pro238, and Ile242 in the TcSpdSyn putrescine-binding site (PDB ID code: 3BWC). When comparing the docking pose with the crystal structure, the binding mode obtained by the docking simulation clearly shows there is no correspondence to the observed binding mode in the crystal structure even though, in both binding modes, the position of the aromatic ring of compound 1 is in the polyamine region of dcSAM. In the TcSpdSyn structure that was used for the docking simulation (PDB ID code: 3BWC), the gatekeeping-loop residue Asp171 is disordered. Thus, interaction with Asp171 was unexpected based on the docking simulation.

The crystal structure analysis of TcSpdSyn with compound 1. (A) The crystal structure of the TcSpdSyn-compound 1 complex. (B) Binding form of compound 1 predicted by docking simulation. (C) Stereo representation of the TcSpdSyn-compound 1 complex structure. The 2Fo-Fc map around compound 1 is shown as a blue mesh (contoured at 1.5σ). (D) Interaction energy analysis of compound 1. The vertical axis shows the interaction energy (kcal/mol) between the ligand and amino acid, and the horizontal axis shows the amino acid residue number. (E) Interaction between compound 1 and Tyr237.
The crystal structure indicates that compound 1 interacts with Asp171 by a salt bridge between the amino group of compound 1 and the carboxylate group of Asp171. To determine whether this interaction meets the geometric requirements of the salt bridge, the distance between the amino nitrogen atom of compound 1 and the δ–O atom of Asp171 was measured. Furthermore, by adding the hydrogen atoms to the crystal structure, we measured the bond angles with respect to the nitrogen, the hydrogen atom attached to the nitrogen, and the δ–O atom of Asp171. We found that the distance and bond angle were 2.71 Å and 162.8°, respectively. Because these values meet the geometric requirements of a salt bridge, we concluded that a salt bridge interaction was present. According to the X-ray crystallography result, the ethylene linked amino group facilitates binding to the TcSpdSyn putrescine-binding site. The IDs of inactive compounds are listed in Supplementary Table 2. In the present study, we confirmed the structures of active compounds using mass spectrometry (MS) and nuclear magnetic resonance (NMR).
Interaction energy analysis
The interaction energy between TcSpdSyn and compound 1 was analyzed by FMO calculation using the crystal structure of TcSpdSyn in complex with compound 1. Figure 3D shows the results of this interaction energy analysis. Compound 1 interacts with Asp171 at the putrescine-binding site (interaction energy value: −16.18 kcal/mol). We also performed FMO calculations using the crystal structure of TcSpdSyn complexed with putrescine and with 4MCHA. The interaction energy values with Asp171 were calculated to be −5.54 and −14.29 kcal/mol, respectively. These significant interaction energy values illustrate interactions with Asp171 and show importance for ligand binding. In addition to the interaction with Asp171, other interactions were confirmed: Gln64 and Tyr73 interact with compound 1 with interaction energy values of −3.32 and −3.25 kcal/mol, respectively. Moreover, Glu201 and Tyr237 interact with compound 1 with interaction energy values of −6.68 and −6.49 kcal/mol, respectively. Figure 3E shows the interaction mode between compound 1 and Tyr237. There appears to stabilization through a π-π interaction between the aromatic ring of compound 1 and the aromatic ring of Tyr237. Thus, the Tyr237 π-electrons play an important role in the binding of compound 1.
Molecular dynamics (MD) analysis of compound interaction
MD simulations were performed for compounds 1–4 and 4MCHA to further investigate their interactions with amino acid residues. For the analysis of compound 1, the complex structure was obtained by X-ray crystallography. For compounds 2–4 the complex structures were obtained by the docking simulation with the compound 1 co-crystal. As a reference, the co-crystal structure of TcSpdSyn and 4MCHA was also subjected to the MD simulation. Figure 4 shows the amino acid residues during the MD simulation. All compounds interact with Asp171 through a salt bridge or hydrogen bond. Moreover, the MD simulation shows that compounds 1–4 interact with other amino acid residues, namely, Gln22, Gln64, Asp168, Gln201, Ser204 and Gly243 suggesting that hit compounds identified in this study have additional interactions that were not predicted by the docking simulations.

TcSpdSyn-ligand interaction analysis by MD simulation. Schemes of detailed ligand atom interactions with the protein residues are presented. Interactions that occur for more than 30.0% of the simulation time in the selected trajectory (0.00 through 20.00 ns) are shown.
Discussion
Using a virtual screening approach, we performed a docking simulation to identify TcSpdSyn inhibitors from among approximately 4,800,000 drug-like compounds. In vitro assays of the compounds selected from the docking simulation were conducted, and the IC50 values of four compounds were determined. As a positive control, we also determined IC50 of 4MCHA against TcSpdSyn (its value is 1.9 uM and is determined by this study.). Furthermore, to clarify the binding modes of the active compounds, X-ray crystallography was performed. The TcSpdSyn-complex 1 structure was determined, whereas attempts to obtain crystal structures of TcSpdSyn in complex with other compounds were unsuccessful. The obtained crystal structure revealed that compound 1 is bound to the TcSpdSyn putrescine-binding site and forms a salt bridge with Asp171 and a van der Waals interaction with Tyr237. Because these two interactions are also observed in the crystal structure of the TcSpdSyn-4MCHA complex, the binding mode of compound 1 is similar to that of 4MCHA. However, despite the conservation of the binding mode, compound 1 is less active than 4MCHA (124 μM vs. 1.9 μM). We believe that there are two possible explanations for the difference in activity. First, there is a high entropic penalty in binding compound 1 due to the fact that compound 1 contains a flexible aminoalkyl side chain, which interacts with Asp171. Second, although 4MCHA clearly forms a CH-π interaction, the phenyl ring of compound 1 is not completely stacked with the aromatic ring of Tyr237, which leads to a weaker π–π interaction. However, we considered compound 1 to be an acceptable starting molecule for optimization because, in many cases of high-throughput screening (HTS) campaigns, the IC50 of hit compounds exceed 10 μM40. In addition, the confirmation rate, which is the ratio of the number of compounds with confirmed enzymatic activity to the number of docking hits, increased to 2.27% because docking simulations were performed; for comparison, the hit rate for HTS is generally <1%27. Moreover, compound 1 is more drug-like than 4MCHA due to the fact that the quantitative estimate of its drug-likeness (QED)41 is greater than that of 4MCHA (compound 1 QED = 0.63 > 4MCHA = 0.47).
According to our FMO analysis results, compound 1 interacts with Gln64, Tyr73, Asp171, Gln201 and Tyr237. Tyr237 interacts with the aromatic ring of compound 1 through a π-π interaction, and Asp171 interacts with the amino group of compound 1 through a salt bridge with the strongest interaction energy (−16.18 kcal/mol). Asp171 belongs to the gatekeeping loop, which is important for substrate binding in the SpdSyn active site28. In the protein structure that we used for the docking simulation (PDB ID code: 3BWC), the gatekeeping loop is disordered because the structure was determined in substrate-free form. However, the gatekeeping loop is found to be ordered in the TcSpdSyn-compound 1 complex structure due to the attraction between Asp171 of this loop and compound 1. We also conducted MD simulations to analyze the interaction modes using compounds 1–4 and 4MCHA. The structures of the TcSpdSyn complex with compounds 2–4 were provided by docking simulations of these compounds using a TcSpdSyn-compound 1 X-ray structure with an ordered gatekeeping loop. The results obtained from MD simulation indicated that, unlike 4MCHA, each compound interacts with Asp171, Gln22, Gln64, Asp168, Gln201, Ser204 and Gly243. In contrast, S-adenosyl-3-thio-1,8-diaminooctane (AdoDATO, PDBID: 2I7C) and 5-(1H-benzimidazol-2-yl)pentan-1-amine (BIPA, PDBID: 4CWA), which are inhibitors of P. falciparum SpdSyn, they bind to both putrescine and dcSAM binding sites in reported co-crystal structures42, 43. Although, compounds 1, 3 and 4 have an amino-alkyl chain like BIPA their amino group should interact with Asp171 through a salt bridge with compound 1 bound to putrescine binding site while compounds 3 and 4 bound to the dcSAM site.
Our results suggest that the docking simulation is a useful method for discovering drug-like inhibitors of TcSpdSyn. Accordingly, we hope to demonstrate that such simulations represent a valuable tool for finding protein inhibitors in general and that the results obtained in this study could facilitate the development of TcSpdSyn-targeted anti-Chagas drugs.
Methods
Target identification
We used the iNTRODB database, which integrates information on the genes, proteins, and compounds related to the three species of parasitic Trypanosoma, to efficiently narrow the range of candidate target molecules11. Using iNTRODB, we identified four candidate target proteins from 9,078 genes using the two-step process shown in Supplementary Figure 1. The present study focuses on SpdSyn.
Protein preparation and docking simulation
The TcSpdSyn structure of 3BWC, which was used as a docking target, was taken from PDB. This structure was subjected to hydrogenation to remove water molecules and optimize the energy by Maestro using the OPLS2005 force field44. Docking simulations were conducted for the putrescine-binding site of the prepared TcSpdSyn structure in the absence of putrescine substrate. While calculating the docking simulation, a grid box with dimensions of 20 × 20 × 20 Å3 was generated to maintain the TcSpdSyn putrescine-binding site. In addition, dcSAM was included as a SpdSyn cofactor.
We used the Glide standard-precision mode45 for docking simulations, and approximately 4,800,000 drug-like compounds, suitable for Lipinski’s rule of five46, from the Namiki Shoji Co., Ltd. (Tokyo, Japan) and Astellas Pharma Inc. (Ibaraki, Japan) in-house libraries were included in the simulation. The mapping table between the compound ID (denoted as supplier ID) and the compounds listed in Table 1 is shown in Supplementary Table 1. All calculations were performed using an HP Proliant SL390s G7 server with an Intel Xeon X5670 2.93 GHz core and five nodes in the TSUBAME2.5 supercomputer located at the Tokyo Institute of Technology.
TcSpdSyn inhibition enzyme assay
The TcSpdSyn inhibition assay was performed essentially as previously27 described. Briefly, the assay was an enzyme-coupled assay incorporating spermidine/spermine N(1)-acetyltransferase 1 (SSAT1). 7-Diethylamino-3-(4-maleimidylphenyl)-4-methylcoumarin (CPM) (cat. D-346, Thermo Fischer Scientific) was used to measure the coenzyme A produced by the SSAT1 reaction. The reaction mixture contained 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.5), 10 μM ethylenediaminetetraacetic acid (EDTA), 0.01% Tween 20, 14.7 nM TcSpdSyn, 50 μM dcSAM, 50 μM putrescine, 15 μM Ac-CoA (acetyl-coenzyme), and 0.83 nM SSAT1 in the presence or absence of the compounds and was incubated at room temperature for 30 min. The concentrations of putrescine and dcSAM were adjusted according to their K m values (data not shown). The fluorescence signal was then detected in a plate reader (Paradigm, Molecular Devices Inc.) with excitation at 405 nm and emission at 530 nm. Each IC50 value was measured using eight different concentrations in quadruplicate. The dose-response curves of the compounds in Table 1 are shown in Supplementary Figure 3. Compounds 1, 3, and 4 were used in their salt forms, whereas compound 2 was used in its free form. The corresponding salts were oxalic acid for compound 1 and HCl for compounds 3 and 4. These compounds were dissolved in dimethyl sulfoxide (DMSO), and the maximum final DMSO concentration in the assays was 1.3%.
Crystallization, data collection, and refinement
X-ray crystallography was carried out essentially as previously described27. Co-crystals of TcSpdSyn in complex with dcSAM and compound 1 were obtained using a sitting-drop vapor diffusion method. Before crystallization, 15 mg/mL TcSpdSyn was mixed with dcSAM and compound 1 at final concentrations of 2 and 5 mM, respectively. The reservoir solution consisted of 100 mM bis-Tris (pH 5.5–6.5), 200 mM ammonium sulfate, and 10–15% (w/v) polyethylene glycol with a molecular weight of 4000 g/mol (PEG 4000). The crystals were transferred into a mother liquor containing 20% (v/v) glycerol as a cryoprotectant and flash frozen in liquid nitrogen. X-ray diffraction data were collected at the Photon Factory AR-NE3A beamline using the robotic sample changer and the automated data collection system47, 48. The structure was solved by molecular replacement with Phaser49 using the apo-structure of TcSpdSyn (PDB ID code: 3BWB) as a search model. After structural refinement using REFMAC50, electron density maps corresponding to dcSAM and compound 1 were clearly obtained and fitted using AFITT (OpenEye Scientific). The final structure was deposited in PDB (PDB ID code: 5B1S).
Interaction energy analysis for SpdSyn–ligand complex
FMO calculation input files were generated using FMOutil Version 2.1(https://staff.aist.go.jp/d.g.fedorov/fmo/fmoutil.html), and calculations were performed for each A-chain monomer using GAMESS51 at the MP2/6–31 G level. Interaction energy analysis was performed using the analytical tool Facio52 on the basis of pair interaction energy decomposition analysis, as proposed by Fedorov et al.53.
MD simulation
Compounds 1–4 and 4MCHA were used in the MD simulations. Complexes of TcSpdSyn and compounds 2–4 were generated by docking simulations using the TcSpdSyn-compound 1 complex structure similarly described at protein preparation and docking simulation section. For compound 1 and 4MCHA, the obtained crystal structures were used in the MD simulation. The simulation system was prepared using Desmond Version 3.5 with default settings. The temperature and pressure of the system were set at 300 K and 1 atm, respectively. The time step and structure sampling intervals were set at 2 fs and 1 ps, respectively. We conducted the simulation under the isothermal-isobaric (NPT) ensemble for 20 ns.
References
Pinto, D. J. C. The treatment of Chagas disease (South American trypanosomiasis). Ann. Intern. Med. 144, 772–774 (2006).
Aufderheide, A. C. et al. A 9,000-year record of Chagas disease. Proc. Natl. Acad. Sci. 101, 2034–2039 (2004).
WHO, Chagas disease (American trypanosomiasis), (2016) (Date of access: 4/5/2016).
Maguire, J. H. Chagas disease-Can we stop the deaths? N. Engl. J. Med. 355, 760–761 (2006).
WHO, The World Health Report, 2002 (World Health Organization, Geneva, 2002).
Inter-American Development Bank: The Millennium Development Goals in Latin America and the Caribbean: Progress, priorities and IDB support for their implementation. Inter-American Development Bank, Washington D.C. 1–70 (2005).
Farrar, J. et al. Manson’s Tropical Infectious Diseases (2013).
The Lancet: Chagas disease – an epidemic that can no longer be ignored. Lancet 368, 619 (2006).
Urbina, J. A. & Docampo, R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 19, 495–501 (2003).
Morillo, C. A. et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 373, 1295–1306 (2015).
Ishida, T. & Akiyama, Y. iNTRODB. (2010) Available at: http://www.bi.cs.titech.ac.jp/introdb/ (Date of access: 4/5/2016).
Aslett, M. et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucl. acid. res. 38, D457–D462 (2010).
Chiba, S. et al. Identification of potential inhibitors based on compound proposal contest: Tyrosine-protein kinase Yes as a target. Scientific Reports 5, doi:10.1038/srep17209 (2015).
Pendeville, H. et al. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol. Cell. Biol. 21, 6549–6558 (2001).
Nishimura, K. et al. Essential role of S-adenosylmethionine decarboxylase in mouse embryonic development. Genes Cells 7, 41–47 (2002).
Gerner, E. W. & Meyskens, F. L. Jr. Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer 4, 781–792, doi:10.1038/nrc1454 (2004).
Janne, J. et al. Genetic approaches to the cellular functions of polyamines in mammals. Eur. J. Biochem. 271, 877–894 (2004).
Ariyanayagam, M. R. et al. Phenotypic analysis of trypanothione synthetase knockdown in the African trypanosome. Biochem. J. 391, 425–432, doi:10.1042/bj20050911 (2005).
Huynh, T. T. et al. Gene knockdown of gamma-glutamylcysteine synthetase by RNAi in the parasitic protozoa Trypanosoma brucei demonstrates that it is an essential enzyme. J. Biol. Chem. 278, 39794–39800, doi:10.1074/jbc.M306306200 (2003).
Krieger, S. et al. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 35, 542–552 (2000).
Li, F. et al. Trypanosoma brucei brucei: characterization of an ODC null bloodstream form mutant and the action of alpha-difluoromethylornithine. Exp. Parasitol. 88, 255–257, doi:10.1006/expr.1998.4237 (1998).
Taylor, M. C. et al. Validation of spermidine synthase as a drug target in African trypanosomes. Biochem. J. 409, 563–569, doi:10.1042/bj20071185 (2008).
Willert, E. K. & Phillips, M. A. Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 4, e1000183, doi:10.1371/journal.ppat.1000183 (2008).
Sergey, K. et al. The crystal structure of spermidine synthase with a multisubstrate adduct inhibitor. Nature Structural Biology 9, 27–31, doi:10.1038/nsb737 (2001).
Wu, M. et al. Structure and mechanism of spermidine synthases. Biochemistry 46, 8331–8339, doi:10.1021/bi602498k (2007).
Pegg, Anthony E., Casero, Jr. & Robert, A. Polyamines-Methods and Protocols. Springer Science+Business Media LLC, New York 3–35 (2011).
Amano, Y. et al. Structural insights into novel inhibition mechanism of Trypanosoma cruzi spermidine synthase. Acta. Crystallogr. D. Biol. Crystallogr. 71, 1879–1889, doi:10.1107/S1399004715013048 (2015).
Lee, M. J., Yang, Y. T., Lin, V. & Huang, H. Site-directed mutations of the gatekeeping loop region affect the activity of Escherichia coli spermidine synthase. Mol. Biotechnol. 54, 572–80, doi:10.1007/s12033-012-9599-3 (2013).
Schneider, G. & Bohm, H. J. Virtual screening and fast automated docking methods. Drug Discov. Today 7, 64–70 (2002).
Grinter, S. Z. & Zou, X. Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules 19, 10150–10176, doi:10.3390/molecules190710150 (2014).
Von Korff, M., Freyss, J. & Sander, T. Comparison of ligand- and structure-based virtual screening on the DUD data set. J. Chem. Inf. Model. 49, 209–231, doi:10.1021/ci800303k (2009).
Kitaura, K. et al. Pair interaction molecular orbital method: an approximate computational method for molecular interactions. Chem. Phys. Lett. 312, 319–324, doi:10.1016/S0009-2614(99)00937-9 (1999).
Yoshino, R. et al. Pharmacophore modeling for anti-chagas drug design using the fragment molecular orbital method. PLoS ONE 10, e0125829, doi:10.1371/journal.pone.0125829 (2015).
Ozawa, T. & Okazaki, K. CH/pi hydrogen bonds determine the selectivity of the Src homology 2 domain to tyrosine phosphotyrosyl peptides: an ab initio fragment molecular orbital study. J. Comput. Chem. 29, 2656–2666, doi:10.1002/jcc.20998 (2008).
Ozawa, T. et al. The importance of CH/pi hydrogen bonds in rational drug design: An ab initio fragment molecular orbital study to leukocyte-specific protein tyrosine (LCK) kinase. Bioorg. Med. Chem. 16, 10311–10318, doi:10.1016/j.bmc.2008.10.041 (2008).
Ozawa, T., Okazaki, K. & Kitaura, K. CH/pi hydrogen bonds play a role in ligand recognition and equilibrium between active and inactive states of the beta2 adrenergic receptor: an ab initio fragment molecular orbital (FMO) study. Bioorg. Med. Chem. 19, 5231–5237, doi:10.1016/j.bmc.2011.07.004 (2011).
Ozawa, T., Okazaki, K. & Kitaura, K. Importance of CH/pi hydrogen bonds in recognition of the core motif in proline-recognition domains: an ab initio fragment molecular orbital study. J. Comput. Chem. 32, 2774–2782, doi:10.1002/jcc.21857 (2011).
Fujimura, K. & Sasabuchi, Y. The role of fluorine atoms in a fluorinated prostaglandin agonist. Chem. Med. Chem. 5, 1254–1257, doi:10.1002/cmdc.201000174 (2010).
Ohno, K. et al. Computational insights into binding of bisphosphates to farnesyl pyrophosphate synthase. Curr. Med. Chem. 18, 220–233 (2011).
Bender, A. et al. Which aspects of HTS are empirically correlated with downstream success? Curr. Opin. Drug Discov. Devel. 11, 327–337 (2008).
Bickerton, G. R. et al. Quantifying the chemical beauty of drugs. Nature Chem. 4, 90–98 (2012).
Janina, S. et al. Binding and Inhibition of Spermidine Synthase from Plasmodium falciparum and Implications for In Vitro Inhibitor Testing. PLoS ONE 11, e0163442, doi:10.1371/journal.pone.0163442 (2016).
Pieter, B. B et al. A novel inhibitor of Plasmodium falciparum spermidine synthase: a twist in the tail. Malaria Journal 14, DOI:10.1186/s12936-015-0572-z (2015).
Banks, J. L. et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 26, 1752–1780, doi:10.1002/jcc.20292 (2005).
Friesner, R. A. et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196, doi:10.1021/jm051256o (2006).
Lipinski, C. A. et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 23, 3–25, doi:10.1016/S0169-409X(96)00423-1 (1997).
Hiraki, M. et al. High-throughput operation of sample-exchange robots with double tongs at the Photon Factory beamlines. J. Synchrotron Radiat. 15, 300–303 (2008).
Hiraki, M. et al. Improvement of an automated protein crystal exchange system PAM for high-throughput data collection. J. Synchrotron Radiat. 20, 890–893 (2013).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D. Biol. Crystallogr. 53, 240–255 (1997).
Schmidt, M. W. et al. General atomic and molecular electronic structure system. Journal of Computational Chemistry 14, 1347–1363, doi:10.1002/jcc.540141112 (1993).
Suenaga, M. Facio: New Computational Chemistry Environment for PC GAMESS. Journal of Computer Chemistry, Japan 4, 25–32, doi:10.2477/jccj.4.25 (2005).
Fedorov, D. G. & Kitaura, K. Pair interaction energy decomposition analysis. J. Comput. Chem. 28, 222–237, doi:10.1002/jcc.20496 (2007).
Acknowledgements
This research was partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) from Japan Agency for Medical Research and Development (AMED), the Research Complex Program “Well-being Research Campus: Creating new values through technological and social innovation” from Japan Science and Technology Agency (JST), and the Japanese Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers 25870222 and 15H02776 (to M.S.), 24240044 (to Y.A.), 15K16082 (to T.I.), 18073004 and 26253025 (to K.K.), 26870119 and 16K19114 (to D.K.I.). The authors thank the anonymous reviewers for their valuable comments and suggestions to improve the quality of the paper.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to this study and article. K.O., T.N., M.O., and M.S. developed the concept and performed the project. T.I., D.K.I., K.K., and Y.A. developed iNTRODB and proposed the target protein. R.Y. and N.Y. conducted virtual screening. Y-S.A., Y.T., and I.N. performed the enzyme assays and X-ray analyses. R.Y., Y.H., and M.S. wrote the main text. All authors approve this version to be published.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoshino, R., Yasuo, N., Hagiwara, Y. et al. In silico, in vitro, X-ray crystallography, and integrated strategies for discovering spermidine synthase inhibitors for Chagas disease. Sci Rep 7, 6666 (2017). https://doi.org/10.1038/s41598-017-06411-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06411-9
This article is cited by
-
End-to-end learning for compound activity prediction based on binding pocket information
BMC Bioinformatics (2021)
-
Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates
Scientific Reports (2020)
-
Molecular Dynamics Simulation reveals the mechanism by which the Influenza Cap-dependent Endonuclease acquires resistance against Baloxavir marboxil
Scientific Reports (2019)
-
A prospective compound screening contest identified broader inhibitors for Sirtuin 1
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
