
Abstract
Rhodoterpenoids A‒C (1‒3), three new rearranged triterpenoids, together with one new biogenetically related compound, rhodoterpenoid D (4), were isolated and efficiently elucidated from Rhododendron latoucheae by high-performance liquid chromatography−mass spectrometry−solid-phase extraction−nuclear magnetic resonance (HPLC‒MS‒SPE‒NMR). Compounds 1 and 2 possess an unprecedented skeleton with a 5/7/6/6/6-fused pentacyclic ring system, while compound 3 contains a unique 6/7/6/6/6-fused pentacyclic carbon backbone. Their structures were determined by extensive spectroscopic methods and electronic circular dichroism (ECD) analyses. Plausible biogenetic pathways for 1‒4 were proposed. Compounds 1 and 4 showed potential activity against herpes simplex virus 1 (HSV-1) with IC50 values of 8.62 and 6.87 μM, respectively.
Similar content being viewed by others
Introduction
The Ericaceae plants have high values in aesthetics and medicine with worldwide distribution. They contain a wide range of chemical components such as flavones1, diterpenes2,3,4, triterpenes5, phenols6, coumarins7, and lignans8, 9 which possess pharmacological activities that include anti-inflammatory10, analgesic2,3,4, anti-oxidant11, anti-bacterial11, anti-HIV12, immunity13, and cytotoxicity14 properties.
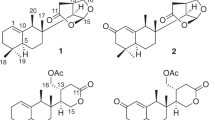
Rhododendron latoucheae Finet et Franch, a plant of the family Ericaceae, is mainly distributed in southern and southwestern mainland China. It has been historically used as a traditional folk medicine for its effects in eliminating phlegm, suppressing cough, activating blood and dissolving stasis15. To date, only two literature articles describing chemical investigations of this plant have been published, and mostly considered the phenols and iridoids16, 17. The previous work carried out by our group over the years led to the isolation of new active terpenoids from plants of the family Ericaceae2,3,4, 18, 19. Therefore, our research on this plant was mainly focused on the attractive terpenoids. A preliminary investigation of this plant indicated that the CH2Cl2-soluble fraction from Rhododendron latoucheae contained many minor triterpenoids that were extremely difficult to obtain by traditional methods of isolation. It is noteworthy that natural products have played an invaluable role in the drug discovery process, and the natural extracts contain many minor constituents, some of which have significant physiological and biological activity20,21,22,23. Here, the application of the hyphenated technique of HPLC‒MS‒SPE‒NMR24, 25, a method that makes the structural analysis of minor components of mixtures feasible, successfully overcame this challenge. Thus, through our further investigation of this plant for the identification of structurally unique and biologically interesting triterpenoids, rhodoterpenoids A‒D (1‒4) (Fig. 1) were isolated by the technology of HPLC‒MS‒SPE‒NMR. Notably, compounds 1 and 2 possess an unprecedented skeleton having a 5/7/6/6/6-fused pentacyclic ring system, while compound 3 contains a unique 6/7/6/6/6-fused pentacyclic carbon backbone. In addition, the new compound 4 obtained from the title plant in the present study was a key precursor before the rearrangement of rings A and B to obtain compound 3. Herein, we report their isolation, structural elucidation, biological evaluation, and possible biogenetic pathways.

Structures of rhodoterpenoids A‒D (1‒4).
Results and Discussion
Rhodoterpenoid A (1) was obtained as a white amorphous powder with the molecular formula C30H48O4, indicating seven degrees of unsaturation (Table 1). The 1H NMR spectrum of 1 [Table 2 and Figure S5 in the Supporting Information (SI)] displayed signals of two secondary methyls at δ H 1.00 (d, J = 6.5 Hz, H3-30) and 1.11 (d, J = 7.1 Hz, H3-29), six tertiary methyls at δ H 0.86 (H3-24), 0.94 (H3-23), 0.99 (H3-25), 1.08 (H3-28), 1.13 (H3-27), and 1.32 (H3-26), two oxygen-bearing methines at δ H 4.04 (dd, J = 9.6 and 8.0 Hz, H-3) and 4.37 (dd, J = 10.9 and 3.7 Hz, H-7), and one olefinic proton at 5.78 (dd, J = 5.9 and 3.4 Hz, H-15). Its 13C NMR spectrum (Table 3 and Figure S6 in the SI) and DEPT experiment revealed eight methyls, seven methylenes, seven methines (one olefinic and two oxygenated), and eight quaternary carbons (one olefinic, one oxygenated, and one ketone). These data suggested a pentacyclic structure and also a triol should be present in 1. The 1H-1H COSY spectrum (Figure S9 in the SI) revealed the presence of the spin-coupling systems shown in bold in Fig. 2. The HMBC correlations (Figure S8 in the SI) from H3-23 to C-3, C-4, C-5, and C-24, from H-1 to C-2, C-5, and C-10, and from H-6a to C-1, C-5, and C-7 allowed the five-membered ring (ring A in Fig. 2) to be defined. Subsequently, the seven-membered ring B fused with ring A was deduced from the HMBC cross-peaks of H3-25 with C-8, C-9, C-10, and C-11, and of H3-26 with C-7, C-8, C-9, and C-14 (Fig. 2). Finally, the common six-membered rings C, D and E were indicated by the HMBC correlations from H-11a to C-8, C-12, C-13, and C-25, from H-15 to C-13, C-16, and C-17, from H3-27 to C-12, C-13, C-14, and C-18, from H3-28 to C-16, C-17, C-18, and C-22, from H3-29 to C-18 and C-20, and from H3-30 to C-19 and C-21 (Fig. 2). Thus, the gross structure of rhodoterpenoid A was elucidated to be 1, which possesses a remarkable 5/7/6/6/6 pentacyclic skeleton.

1H‒1H COSY and key HMBC correlations of (1).
The relative configuration of 1 was elucidated by nuclear Overhauser effect (NOE) experiments (Fig. 3 and Figure S10 in the SI). The NOE correlations of H-3/H3-23, H-1/H3-24, H-1/H-6b, H-6a/H-7/H-11a, and H-7/H3-27/H-16a suggested that H-3, H-7, H3-23, H3-27 and OH-5 were cofacial and α-oriented. In addition, the NOE correlations of H-1/H3-26/H3-25, H3-26/H-15, H-16b/H3-28/H-20, H-19/H3-27, and H3-28/H-18/H3-29 confirmed that H-1, H-18, H-20, H3-24, H3-25, H3-26, H3-28 and H3-29 were on the same side with a β -direction.

Key NOE correlations for (1) and (2).
Based on the above results, there were only two possible structures for 1, with absolute configurations of 1a (1 S, 3 S, 5 S, 7 S, 8 S, 9 R, 13 S, 17 R, 18 R, 19 S, 20 R) and 1b (1 R, 3 R, 5 R, 7 R, 8 R, 9 S, 13 R, 17 S, 18 S, 19 R, 20 S). Thus, the electronic circular dichroism (ECD) spectra of 1a and its enantiomer 1b were calculated using TDDFT. The experimental ECD spectrum of 1 was in good agreement with the calculated ECD of 1a and was the opposite of 1b (Fig. 4A), which suggested an absolute configuration of 1 S, 3 S, 5 S, 7 S, 8 S, 9 R, 13 S, 17 R, 18 R, 19 S and 20 R for compound 1. This absolute configuration of 1 was substantiated by the olefin octant rule (Fig. 4B). The experimental ECD spectrum of 1 showed a positive Cotton effect near 200 nm, corresponding to the π → π* electronic transition of the Δ14 double bond26, 27.

(A) Experimental ECD spectrum of 1, using TDDFT at the B3LYP/6-31 G(d) level in MeOH calculated ECD spectra of 1a and 1b. (B) Application for the olefin octant rule of 1 (rear octants viewed along Y-axis).
Rhodoterpenoid B (2) was assigned a molecular formula C30H46O5 on the basis of its (+)-HRESIMS data (Table 1). A comparison of the 1H and 13C NMR data of 2 with those of 1 (Tables 2 and 3) revealed that they possessed the same carbon skeleton and implied a C-16 carbonyl in 2 instead of a methylene in 1 (C-16: δ C 208.9 for 2, δ C 42.9 for 1). This deduction was confirmed by the HMBC correlations (Figure S18 in the SI) of H3-28 with C-16, of H-15 with C-16, of H-22 with C-16, and of H-18 with C-16. The key NOE correlations (Fig. 3 and Figure S20 in the SI) of H-3/H3-23, H3-24/H-1/H3-26/H-6, H-7/H-11a, H-7/H3-27/H-19, H-15/H3-26/H3-25, and H-20/H3-28/H3-29 confirmed that the relative configuration of 2 was absolutely identical to that of 1.
The absolute configuration of compound 2, was established identical as that of 1 by the same procedure of ECD spectra calculation of both enantiomers. The experimental ECD spectrum of 2 agreed well with the one calculated for 2a (1 S, 3 S, 5 S, 7 S, 8 S, 9 R, 13 S, 17 S, 18 S, 19 S, 20 R) and was the opposite of 2b (Fig. 5A). The absolute configuration of compound 2 was further substantiated by applying the helicity rule28, 29 for the α,β-unsaturated ketone with a negative Cotton effect at 253 nm (Fig. 5B).

(A) Experimental ECD spectrum of 2, using TDDFT at the B3LYP/6-31 G(d) level in MeOH calculated ECD spectra of 2a and 2b. (B) Application for the helicity rule of 2.
Rhodoterpenoid C (3) was assigned a molecular formula of C30H44O5 by its HRESIMS data (Table 1). The 1H NMR spectrum of 3 (Table 2 and Figure S25 in the SI) showed signals for two secondary methyls at δ H 1.06(d, J = 6.1 Hz, H3-30) and 1.14 (d, J = 6.6 Hz, H3-29), six tertiary methyls at δ H 0.65 (H3-27), 0.77 (H3-26), 1.17 (H3-28), 1.22 (H3-24), 1.24 (H3-23), and 1.38 (H3-25), three oxygen-bearing methines at δ H 3.44 (ddd, J = 11.7, 9.8 and 6.6 Hz, H-21), 4.01 (dd, J = 11.1 and 5.7 Hz, H-15), and 4.41 (dd, J = 3.8 and 2.5 Hz, H-6), and two olefinic proton at 5.64 (brs, H-7) and 5.75 (dd, J = 3.8 and 3.7 Hz, H-11). Its 13C NMR spectrum (Table 3 and Figure S26 in the SI) and DEPT experiment disclosed eight methyls, four methylenes, nine methines (two olefinic and three oxygenated), and nine quaternary carbons (two olefinic and two ketones). Thus, compound 3 was also a pentacyclic triterpenoid and four aditional degrees of unsaturation described in the above data. The 1H-1H COSY spectrum (Figure S29 in the SI) suggested the presence of the spin-coupling systems shown in bold in Fig. 6. The HMBC correlations (Figure S28 in the SI) from H3-23 to C-3, C-4, C-5, and C-24, from H3-25 to C-1, C-5, C-9, and C-10, and from H-6 to C-1, C-2, C-3, C-7, and C-8 allowed the six-membered carbon ring (ring A in Fig. 6) to be defined. Then, the seven-membered ring B sharing C-1, C-2, and C-10 with ring A was deduced from the HMBC cross-peaks of H-7 with C-9 and C-14 and of H-11 with C-8 and C-10 (Fig. 6). Finally, the common six-membered rings C, D and E were indicated by the HMBC correlations from H3-26 to C-8, C-13, C-14, and C-15, from H3-27 to C-12, C-13, C-14, and C-18, from H3-28 to C-16, C-17, C-18, and C-22, from H3-29 to C-18, and C-20, and from H3-30 to C-19, and C-21 (Fig. 6). Therefore, the gross structure of rhodoterpenoid C was elucidated to be 3, which contains a unique 6/7/6/6/6-fused pentacyclic carbon backbone. The relative configuration of 3 was deduced from NOE experiments. The NOE correlations (Fig. 6 and Figure S30 in the SI) of H3-23/H-1a/H-2, H-1a/H3-25/H-11, H-1b/H-6/H3-27/H-21/H3-30, and H-7/H-15/H3-27 showed that H-2, H-6, H-15, H-21, H3-23, H3-25 and H3-27 were cofacial and α-oriented. In addition, the NOE correlations (Fig. 6) of H3-26/H-18/H3-28 and H-18/H3-29/H-20 indicated that H-18, H-20, H3-26, H3-28, and H3-29 were on the same side with a β -direction.

Selected 1H-1H COSY, HMBC, and NOE correlations for (3) and (4).
As previously, there were only two possible structures for 3 (3a and 3b), and the absolute configuration was established via experimental and calculated ECD (after failed attempts to obtain a single crystal of 3). Again, the experimental ECD spectrum of 3 correlated fairly well with the calculated ECD of 3a and was the opposite of that of 3b (Fig. 7A), which suggested an absolute configuration of 2 S, 6 R, 10 R, 13 S, 14 S, 15 R, 17 R, 18 R, 19 R, 20 S and 21 S for compound 3. The negative Cotton effect at 247 nm (Fig. 7B) by applying the helicity rule for non-planar transoid dienes30 further demonstrated this absolute configuration of 3.

(A) Experimental ECD spectrum of 3, using TDDFT at the B3LYP/6-31 G(d) level in MeOH calculated ECD spectra of 3a and 3b. (B) Application for the helicity rule of 3.
Rhodoterpenoid D (4) was assigned a molecular formula of C30H44O3 by its HRESIMS data (Table 1). Extensive analysis of 1D (Tables 2 and 3) and 2D NMR spectra revealed that 4 possessed a bauerene skeleton. The proposed structure of 4 was fully determined by its HSQC, 1H−1H COSY, and HMBC spectra (Fig. 6). The key NOE correlations (Fig. 6) of H-15/H3-27/H-21 confirmed that H-15 and H-21 were on the same side with a α-direction, and its absolute configuration was finally deduced as 10 R, 13 S, 14 S, 15 R, 17 R, 18 R, 19 R, 20 S and 21 S by applying the octant rule for the saturated ketone31. The plane projection of optimized conformation of 4 with the above absolute configuration on the rear octants showed the negative sign, which agreed well with the negative Cotton effect at 312 nm in the experimental ECD spectrum.
A plausible biogenetic pathway for rhodoterpenoids A‒D (1‒4) was proposed as shown in Fig. 8. Rhodoterpenoids A‒C (1‒3) are novel rearranged triterpenoids that may be derived from α-amyrin, which is a common triterpene occurring in natural plant populations32. As shown, α-amyrin underwent several dehydrogenations, hydrogenations, oxidations, methyl shifts, ring-opening reactions by double-bond oxidation and cyclizations to afford compounds 1‒3. It is noteworthy that the isolation of new compound 4, which was a key precursor before the rearrangement of rings A and B to obtain compound 3, rationalized the proposed biogenetic pathway for compound 3.

Plausible Biogenetic Pathway of (1‒4).
The compounds were tested for their anti-inflammatory and anti-virus activities, because some studies have shown that the pentacyclic triterpenoids have these activities33,34,35,36. Compounds 1‒4 were evaluated in vitro for anti-inflammatory activity using the measurement of the inhibition of lipopolysaccharide (LPS)-induced TNF-α production in RAW264.7 cells by enzyme-linked immunosorbent assay37 with dexamethasone as a positive control. Compounds 2‒4 showed relatively weak inhibitory effects, with inhibition of TNF-α production at 29%, 7.9% and 30%, respectively, at a concentration of 10 μM. Because of the scarce amounts obtained of compound 3, only compounds 1, 2 and 4 were tested for antiviral [herpes simplex virus-1 (HSV-1)] activity. Compounds 1 and 4 showed potential activity against HSV-1, with IC50 values of 8.62 and 6.87 μM, respectively, and compound 2 was inactive (Table 4). The above data implied that replacement of the methylene at C-16 in 1 with a ketone carbon dramatically decreased anti-HSV-1 activity in 2.
In summary, four new triterpenoids, including two with an unprecedented 5/7/6/6/6-fused pentacyclic ring system and one with a unique 6/7/6/6/6-fused pentacyclic carbon backbone, were rapidly obtained and efficiently elucidated from Rhododendron latoucheae by HPLC‒MS‒SPE‒NMR. Their absolute configurations were determined by ECD analyses combined with experimental rules26,27,28,29,30,31 after unsuccessful attempts to obtain single crystals of 1‒3. It is significant that the minor novel compounds were isolated and efficiently elucidated by the hyphenated technique of HPLC‒MS‒SPE‒NMR. These newly discovered compounds not only provide new challenges and opportunities for synthesis and biological evaluation but also prompt us to pay more attention to the triterpenes of the Ericaceae plants.
Materials and Methods
General experimental procedures
Optical rotations were measured with a JASCO P-2000 automatic digital polarimeter. UV spectra were measured on a JASCO V650 spectrophotometer. CD spectra were recorded on a JASCO-815 CD spectrometer. IR spectra were measured on a Nicolet 5700 FT-IR microscope instrument. HPLC–MS–SPE–NMR were carried out by using a chromatographic separation system consisted of an Agilent 1260 series HPLC with an in-line solvent degasser, quaternary pump, auto-sampler, column compartment with thermostat, and a diode array detector. The chromatographic separation was carried out using a YMC-Pack Pro C18 column (4.6 mm × 250 mm, 5μm), and the column temperature was maintained at 40 °C. ESIMS were performed using a Bruker micrOTOF-Q II, while NMR spectra were obtained on a Bruker AVANCE III HD 600 MHz spectrometer except for that the NOE spectra of compounds 1 and 4 were measured on an Agilent-NMR-vnmrs 600 spectrometer. Chemical shifts are given in δ (ppm) with the solvent (CD3OD; δ H 3.31; δ C 49.0) peaks used as references. (+)-HRESIMS data of compounds 1–3 were recorded using an Agilent 6520 Accurate-Mass Q-TOF LC/MS spectrometer, and (+)-HRESIMS data of compound 4 was performed on a Bruker micrOTOF-Q II. Polyamide resin (30−60 mesh, Jiangsu Linjiang Chemical Reagents Factory, Linjiang, China), macroporous resin (D101 type, Chemical Plant of Nankai University, Nankai, China), MCI gel (Mitsubishi Chemical Corporation), Sephadex LH-20 (GE Chemical Corporation), Silica gel (200–300 mesh, Qingdao Marine Chemical Factory, China), and ODS (50 μm, Merck, Germany) were used for column chromatography (CC). TLC was carried out with glass precoated Silica gel GF254 plates (Qingdao Marine Chemical Factory, China). Spots were visualized by spraying with 10% H2SO4 in EtOH followed by heating.
Plant material
Twigs and leaves of Rhododendron latoucheae were collected from Zhangjiajie, Hunan Province, People’s Republic of China, in October 2014 and identified by Prof. Lin Ma of the Chinese Academy of Medical Sciences and Peking Union Medical College. A voucher specimen (ID-22815) was deposited in the herbarium at the Department of Medicinal Plants, Institute of Materia Medica, Chinese Academy of Medical Sciences.
Extraction and isolation
The air-dried twigs and leaves of Rhododendron latoucheae (107 kg) were extracted with 95% EtOH/H2O (2 h × 3; 10 L/Kg) under reflux conditions. The crude extract (6000 g) was suspended in 30 L of H2O and then partitioned with petroleum ether, CH2Cl2, EtOAc and n-butanol (3 × 30 L). The CH2Cl2-soluble fraction (500 g) was subjected to a MCI gel column eluted with MeOH‒H2O (90:10, 100:0 v/v). The 90% MeOH fraction (350 g) was then further separated over a silica gel column and eluted in a gradient of petroleum ether/(Me)2CO (50:1, 30:1, 20:1, 10:1, 5:1, 1:1 v/v) to afford Fr.1‒Fr.7. Fr.4 (40 g) was applied to a Sephadex LH-20 column (petroleum ether/CH2Cl2/MeOH, 5:5:1) to obtain Fr.4-1 (25 g), which was further resolved on a Sephadex LH-20 column eluted with MeOH to get four subfractions (Fr.4-1-1‒Fr.4-1-4). Fr.4-1-3 (20 g) was subjected to MCI gel column eluted using a gradient of MeOH-H2O (30:70, 60:40, 70:30, 80:20, 100:0 v/v) to produce eleven subfractions (Fr.4-1-3 A‒Fr.4-1-3 K). Fr.4-1-3E (800 mg) was chromatographed over Sephadex LH-20 and ODS gel columns to produce five subfractions (Fr.4-1-3E4-1‒Fr.4-1-3E4-5). Fr.4-1-3E4-5 (20 mg) was purified by HPLC–MS–SPE–NMR with MeCN/H2O/TFA (45:55:0.055, v/v/v, 1.0 mL/min) to yield compound 3 (1.5 mg, tR = 17.7 min) and 2 (2 mg, tR = 20.1 min). Fr.4-1-3I (5 g) was subjected to silica gel, Sephadex LH-20 and ODS gel columns to produce Fr.4-1-3I4-1-5 (35 mg), which was further purified by HPLC–MS–SPE–NMR with MeOH/H2O/TFA (75:25:0.025, v/v/v, 1.0 mL/min) to afford compound 1 (2 mg, tR = 50.3 min). Similarly, Fr.4-1-3 H (1 g) was subjected to Sephadex LH-20 and ODS gel columns to produce Fr.4-1-3H2-3 (20 mg), which was subsequently purified by HPLC–MS–SPE–NMR with MeCN/H2O/TFA (50:50:0.05, v/v/v, 1.0 mL/min) to obtain compound 4 (2 mg, tR = 43.3 min).
Rhodoterpenoid A (1) white amorphous powder; \({[\alpha ]}_{D}^{20}\) + 38.9 (c 0.1, MeOH); UV (MeOH) λ max (log ε): 203 (3.68) nm; CD (MeOH) max (Δε) 290 (+0.52) nm; IR (KBr) νmax: 3398, 2927, 2868, 1685, 1464, 1380, 1079, 1050, 1024, 976, 883, 837, 803, 724 cm−1; 1H NMR (CD3OD, 600 MHz) data, see Table 2; 13C NMR (CD3OD, 150 MHz) data, see Table 3; (+)-HRESIMS m/z 495.3438 [M + Na]+ (calcd for C30H48O4Na, 495.3445).
Rhodoterpenoid B (2). white amorphous powder; \({[\alpha ]}_{D}^{20}\) + 19.6 (c 0.13, MeOH); UV (MeOH) λ max (log ε): 244 (3.39) nm; CD (MeOH) max (Δε) 225 (+0.85), 253 (−0.5), 292 (+0.47), 334 (+0.54) nm; IR (KBr) νmax: 3394, 2957, 2933, 2872, 1686, 1640, 1459, 1380, 1307, 1249, 1182, 1094, 1071, 1049, 1027, 975, 914, 838, 802, 711, 630 cm−1; 1H NMR (CD3OD, 600 MHz) data, see Table 2; 13C NMR (CD3OD, 150 MHz) data, see Table 3; (+)-HRESIMS m/z 487.3428 [M + H]+ (calcd for C30H47O5, 487.3418).
Rhodoterpenoid C (3). white powder; \({[\alpha ]}_{D}^{20}\) – 79.9 (c 0.04, MeOH); UV (MeOH) λ max (log ε): 236 (3.56), 203 (3.56) nm; CD (MeOH) max (Δε) 247 (−6.11), 305 (−1.45) nm; IR (KBr) νmax: 3423, 2931, 2871, 1687, 1464, 1378, 1270, 1050, 1016, 903, 639 cm−1; 1H NMR (CD3OD, 600 MHz) data, see Table 2; 13C NMR (CD3OD, 150 MHz) data, see Table 3; (+)-HRESIMS m/z 507.3081 [M + Na]+ (calcd for C30H44O5Na, 507.3081).
Rhodoterpenoid D (4). colorless oil; \({[\alpha ]}_{D}^{20}\) – 121.8 (c 0.07, MeOH); UV (MeOH) λ max (log ε): 318 (3.46), 306 (3.44), 206 (3.74) nm; CD (MeOH) max (Δε) 208 (+10.08), 312 (−6.81) nm; IR (KBr) νmax: 3428, 2957, 1709, 1462, 1380, 1249,1077, 1040, 1014, 930, 905, 661 cm−1; 1H NMR (CD3OD, 600 MHz) data, see Table 2; 13C NMR (CD3OD, 150 MHz) data, see Table 3; (+)-HRESIMS m/z 453.3357 [M + H]+ (calcd for C30H45O3, 453.3363).
Methodology for ECD calculation and experimental rules of 1–3
Conformational analysis of 1–3 was performed using the MMFF94 molecular mechanics force field by MOE software and the conformers were optimized at B3LYP/6-31 G(d) level. The 50 lowest electronic transitions for the optimized conformations of 1–3 in MeOH were calculated using TDDFT method, and their theoretical ECD spectra were afforded by a Gaussian function with a half-bandwidth of 0.35 eV. The application of experimental rules for compounds 1–4 were based on their only optimized conformations obtained by the above method.
Anti-inflammatory assay
RAW264.7 cells were cultured in 96-well plates (1 × 104 cell mL−1) and preincubated with compounds for 1 h, followed by a further 12 h treatment with LPS for measurement of TNF-α. TNF-α content in the culture medium were measured by ELISA using anti-mouse TNF-α antibodies and a biotinylated secondary antibody, according to the manufacturer’s instructions. ELISA kit was obtained from Invitrogen by Thermo Fisher Scientific(Catalog numbr:88-7324). The optical density of each well was measured at 450 nm with an ELISA reader (Synergy H1, BioTeck, VT, USA). The RAW264.7 cells were obtained from the American Type Culture Collection (ATCC).
Antiviral assays
African green monkey kidney cells (Vero) were from the Institute of Virology, Chinese Academy of Preventive Medicine38. Herpes simplex virus-1 (HSV-1 F strain VR 733) were obtained from the American Type Culture Collection (ATCC).
Cytotoxic assay
The cytotoxicity of compounds 1, 2, and 4 in the presence of Vero cells were monitored by cytopathic effect (CPE). Vero cells (2.5 × 104/well) were plated into a 96-well plate. A total of 24 h later, the monolayer cells were incubated with various concentrations of test compounds. After 48 h of culture at 37 °C and 5% CO2 in a carbon-dioxide incubator, the cells were monitored by CPE. Median toxic concentration (TC50) was calculated by Reed and Muench analyses.
Anti-HSV-1 assay
The anti-HSV-1 activity of the compounds 1, 2, and 4 was assayed by the CPE inhibition method. Vero cells (2.5 × 104cells/well) were plated into 96-well culture plates for an incubation period of 24 h. The cells were infected with 100 μL of HSV-1 at 100TCID50 for 2 h at 37 °C. Then, various concentrations of the test compounds were added after removed the medium. Viral cytopathic effect (CPE) was observed when the CPE of the control group cells reached a value of 4+. Each experiment was performed in triplicate at least three separate times. The IC50 value is defined as the minimal concentration of inhibitor required to inhibit 50% of the CPE, as determined by the Reed and Muench method. The selectivity index was calculated as the ratio of TC50/IC50.
References
Zhou, W. et al. Phytochemical studies of korean endangered plants: a new flavone from Rhododendron brachycarpum. G. Don. Bull. Korean Chem. Soc. 44, 2535–2538 (2013).
Li, Y. et al. Rhodomollins A and B, two diterpenoids with an unprecedented backbone from the fruits of Rhododendron molle. Sci. Rep. 6, 36752, doi:10.1038/srep36752 (2016).
Li, Y. et al. Mollanol A, a diterpenoid with a new C-nor-D-homograyanane skeleton from the fruits of Rhododendron molle. Org. Lett. 16, 4320–4323 (2014).
Li, Y. et al. Mollolide A, a diterpenoid with a new 1,10:2,3-disecograyanane skeleton from the roots of Rhododendron molle. Org. Lett. 15, 3074–3077 (2013).
Takahashi, H., Hirata, S., Minami, H. & Fukuyama, Y. Triterpene and flavanone glycoside from Rhododendron simsii. Phytochemistry 56, 875–879 (2001).
Yao, G. M., Wang, Y. B., Wang, L. Q. & Qin, G. W. Phenolic glucosides from the leaves of Pieris japonica. Acta Pharm. Sin. 43, 284–290 (2008).
Ahmad, V. U. et al. Tyrosinase inhibitors from Rhododendron collettianum and their structure-activity relationship (SAR) studies. Chem. Pharm. Bull. 52, 1458–1461 (2004).
Kashima, K. et al. five new lignans from Lyonia ovalifolia. Chem. Pharm. Bull. 58, 191–194 (2010).
Guo, Q. et al. Five new compounds from Rhododendron mariae Hance. J. Asian Nat. Prod. Res. 16, 1–10 (2014).
Ku, S. K. et al. Anti-inflammatory effects of hyperoside in human endothelial cells and in mice. Inflammation 38, 784–799 (2015).
Wang, C. M. et al. Structure elucidation of procyanidins isolated from Rhododendron formosanum and their anti-oxidative and anti-bacterial activities. Molecules 20, 12787–12803 (2015).
Kashiwada, Y. et al. Isolation of rhododaurichromanic acid B and the anti-HIV principles rhododaurichromanic acid A and rhododaurichromenic acid from Rhododendron dauricum. Tetrahedron 57, 1559–1563 (2001).
Liu, Y. Z. et al. Immunomodulatory effects of proanthocyanidin A-1 derived in vitro from Rhododendron spiciferum. Fitoterapia 81, 108–114 (2010).
Louis, A., Petereit, F., Lechtenberg, M., Deters, A. & Hensel, A. Phytochemical characterization of Rhododendron ferrugineum and in vitro assessment of an aqueous extract on cell toxicity. Planta Med. 76, 1550–1557 (2010).
The China Medicinal Materials Group. Main Record of Resource of Chinese Material Medicine in China (Eds. Zhang, H. Y., Zhang, Z. Y.) 892 (Zhang, Z. Y., 1994).
Fan, C. Q., Yang, G. J., Zhao, W. M., Ding, B. Y. & Qin, G. W. Phenolic components from Rhododendron latoueheae. Chin. Chem. Let. 10, 567–570 (1999).
Fan, C. Q., Zhao, W. M., Ding, B. Y. & Qin, G. W. Constituents from the leaves of Rhododendron latoucheae. Fitoterapia 72, 449–452 (2001).
Niu, C. S. et al. Analgesic diterpenoids from the twigs of Pieris formosa. Tetrahedron 72, 44–49 (2016).
Li, Y. et al. Antinociceptive grayanoids from the roots of Rhododendron molle. J. Nat. Prod. 78, 2887–2895 (2015).
Djeddi, S. et al. Minor sesquiterpene lactones from Centaurea pullata and their antimicrobial activity. J. Nat. Prod. 70, 1796–1799 (2007).
Chen, M. et al. Enantiomers of an indole alkaloid containing unusual dihydrothiopyran and 1,2,4-thiadiazole rings from the root of Isatis indigotica. Org. Lett. 14, 5668–5671 (2012).
Tian, Y. et al. A minor diterpenoid with a new 6/5/7/3 fused-ring skeleton from Euphorbia micractina. Org. Lett. 16, 3950–3953 (2014).
Tang, Z. H. et al. Antiviral spirotriscoumarins A and B: two pairs of oligomeric coumarin enantiomers with a spirodienone-sesquiterpene skeleton from Toddalia asiatica. Org. Lett. 18, 5146–5149 (2016).
Liu, H. et al. Identification of three novel polyphenolic compounds, origanine A–C, with unique skeleton from Origanum vulgare L. using the hyphenated LC-DAD-SPE-NMR/MS methods. J. Agric. Food Chem. 60, 129–35 (2012).
Castro, A., Moco, S., Coll, J. & Vervoort, J. LC-MS-SPE-NMR for the isolation and characterization of neo-Clerodane diterpenoids from Teucrium luteum subsp. flavovirens. J. Nat. Prod. 73, 962–965 (2010).
Scott, A. I. & Wrixon, A. D. A symmetry rule for chiral olefins. Tetrahedron 26, 3695–3715 (1970).
Scott, A. I. & Wrixon, A. D. Stereochemistry of olefins—IX*: correlation of Mills’ and Brewster’s rules with the Cotton effects of cyclic olefins. Tetrahedron 27, 4787–4819 (1971).
Gawronski, J. Circular dichroism and stereochemistry of chiral conjugated cyclohexenones. Tetrahedron 38, 3–26 (1982).
Snatzke, G. A. Circular dichroism and optical rotatory dispersion—principles and application to the investigation of the stereochemistry of natural products. Angew. Chem., Int. Ed. Engl. 7, 14–25 (1968).
Charney, E., Ziffer, H. & Weiss, U. Optical activity of non-planar conjugated dienes-II. Tetrahedron 21, 3121–3126 (1965).
Lightner, D. A. Circular Dichroism, Principles and Applications (Eds. Nakanishi, K., Berova, N., and Woody, R. W.) 259–299 (1994).
Rao, V. Phytochemicals-A Global Perspective of Their Role in Nutrition and Health, (Ed. Rao, V.), 487–502 (Vazquez, L. H., 2012).
Ángel, L., Solomon, Á. & Francisco, H. P. Inhibitory effects of lupene-derived pentacyclic triterpenoids from Bursera simaruba on HSV-1 and HSV-2 in vitro replication. Nat. Prod. Res. 29, 2322–2327 (2015).
Verano, J., González-Trujano, M. E., Déciga-Campos, M., Ventura-Martínez, R. & Pellicer, F. Ursolic acid from Agastache mexicana aerial parts produces antinociceptive activity involving TRPV1 receptors, cGMP and a serotonergic synergism. Pharmacol. Biochem. Behav. 110, 255–264 (2013).
Reyes, C. P. et al. Activity of lupane triterpenoids from Maytenus species as inhibitors of nitric oxide and prostaglandin E2. Bioorg. Med. Chem. 14, 1573–1579 (2006).
Gong, Y. et al. The synergistic effects of betulin with acyclovir against herpes simplex viruses. Antivir. Res. 64, 127–130 (2004).
Engvall, E. & Perlmann, P. Enzyme-linked immunosorbent assay (ELISA), quantitative assay of immunoglobulin G. Immunochemistry 8, 871–874 (1971).
Li, Y. P. et al. Synthesis and biological evaluation of heat-shock protein 90 inhibitors: geldanamycin derivatives with broad antiviral activities. Antiviral Chem. Chemother. 20, 259–268 (2010).
Acknowledgements
This work was supported by grants from the CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-010) and the National Natural Science Foundation of China (No. 21572274). The authors are grateful to the Department of Instrumental Analysis at our institute for the spectroscopic measurements.
Author information
Authors and Affiliations
Contributions
S.S.Y. designed the study and supervised the project. F.L., Y.N.W., Y.L., S.G.M., J.Q., Y.B.L., C.S.N., and Z.H.T. performed the isolation and structure analysis of the compounds. T.T.Z. and Y.H.L. performed and analyzed the bioassay. L.L. finished the ECD computation. S.S.Y. and F.L. wrote the paper. All authors contributed to the discussion and interpretation of the results.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, F., Wang, YN., Li, Y. et al. Rhodoterpenoids A‒C, Three New Rearranged Triterpenoids from Rhododendron latoucheae by HPLC‒MS‒SPE‒NMR. Sci Rep 7, 7944 (2017). https://doi.org/10.1038/s41598-017-06320-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06320-x
This article is cited by
-
Anti-viral triterpenes: a review
Phytochemistry Reviews (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
