
Abstract
Although much progress has been made in the study of cell wall biosynthetic genes in the model filamentous fungus Aspergillus nidulans, there are still targets awaiting characterization. An example is the gene celA (ANIA_08444) encoding a putative mixed linkage glucan synthase. To characterize the role of celA, we deleted it in A. nidulans, analyzed the phenotype of the mycelium and performed RNA-Seq. The strain shows a very strong phenotype, namely “balloons” along the hyphae and aberrant conidiophores, as well as an altered susceptibility to cell wall drugs. These data suggest a potential role of the gene in cell wall-related processes. The Gene Ontology term Enrichment analysis shows increased expression of secondary metabolite biosynthetic genes (sterigmatocystin in particular) in the deleted strain. Our results show that the deletion of celA triggers a strong phenotype reminiscent of cell wall-related aberrations and the upregulation of some secondary metabolite gene clusters in A. nidulans.
Similar content being viewed by others

Introduction
The cell wall is a structure involved in important stages of fungal growth and morphogenesis. Several studies in the literature have shown how perturbations at the cell wall-level trigger dramatic effects on growth1. Despite the importance of fungal cell walls and despite the great advances made in the field, there are still missing pieces in our understanding of cell wall dynamics in filamentous fungi. Some cell wall biosynthetic genes, for example, are still uncharacterized (a detailed inventory of Aspergillus nidulans cell wall-related genes has been previously published2). The chief polysaccharides in the cell wall of the model organism A. nidulans are β-glucans (β-1,3-, β-(1,3;1,4)- and β-1,6-glucans), chitin and α-1,3-glucans. No characterization is yet available for the proposed mixed linkage glucan synthase gene celA ANIA_084442, while much is known about the chitin and α-1,3-glucan biosynthetic genes in A. nidulans 3,4,5,6,7,8. In A. nidulans 6 chitin synthase genes have for instance been identified which belong to classes I to VI3,4,5,6,7. chsC and chsA were proven to play an important role in hyphal wall integrity and conidiophore development4, while chsB and chsD are involved in hyphal growth and conidiation5. Interestingly two of the identified chitin synthases possess a myosin-motor head domain7, which was shown to be important for the interaction with the cytoskeleton and to play a role for the correct delivery and insertion in the plasma membrane.
Recently, a study on A. fumigatus has characterized a protein with significant similarity to ANIA_08444 termed Tft1, an enzyme shown to be responsible for the production of β-(1,3;1,4)-glucans in this organism9.
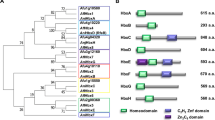
The protein encoded by celA is distantly related to plant cellulose synthase-like proteins (CSLs; Fig. 1), it is orthologous to the characterized A. fumigatus mixed-linkage glucan synthase Tft19, whose deletion causes no obvious phenotype in A. fumigatus, except for a modest increase in virulence.

Neighbor-joining tree (bootstraps = 1000) of CelA, its fungal and plant orthologs. Accession numbers are indicated in the tree.
To characterize the role of celA in the growth and development of the model filamentous fungus A. nidulans, we here sought to provide transcriptomic data of a strain which lacks the gene celA (the strain is hereafter referred to as celA∆).
In the present study, we provide experimental evidence for a relationship existing between the deletion of the gene celA and the upregulation of genes involved in the production of secondary metabolites (SM). We propose that celA partakes in a mechanism regulating SM production in A. nidulans.
Results and Discussion
Deletion of celA causes a strong phenotype in A. nidulans
Transformation of the SAA.111 recipient strain with the replacement cassette containing the argB selectable marker yielded three independent transformants; however, subsequent growth cycles of the transformants from conidia on selective medium resulted in one single transformant as capable of stable growth under arginine deprivation. This celAΔ strain was analyzed and found to have a single and correct argB marker insertion replacing celA in the genome (Suppl. Figure 1a; for a schematic representation of the EcoRI restriction map in the wild-type and celAΔ, see Suppl. Figure 1b). The diagnostic PCRs highlighted the expected shift in MW caused by deletion of celA and insertion of the argB marker (Suppl. Figure 1c and d).
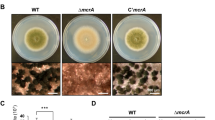
When grown on solid medium, the celAΔ strain showed a macrophenotype: besides growing more slowly, it developed a colony with an outer sector devoid of pigments, as compared to the control (Fig. 2a). The celAΔ strain showed decreased sensitivity to the cell wall drugs Congo Red (CR) and dichlobenil (DCB) (Fig. 2b). Microscopic observations of the hyphae in the celAΔ strain revealed frequently swollen regions and “balloons”; at higher magnifications these structures appeared vacuolated (Fig. 2c). Additionally, several conidiophores retrieved from plate cultures were aberrant, since they showed bifurcations and hyphae-like metulae (Fig. 2c insets). It is known that A. nidulans strains lacking specific chitin synthase genes of class V and VI also show a “balloon” phenotype7, which is indicative of a weakened cell wall. The results here obtained therefore suggest a potential implication of celA in cell wall-related processes; however its actual role in cell wall biosynthesis awaits experimental validation. As the deletion of genes triggering phenotypes reminiscent of those observed in celAΔ is often accompanied by compensatory alterations in expression of genes involved in cell wall biosynthesis10, we tested some of their representatives. As an example, genes involved in chitin and β-1,3-glucan synthesis (Fig. 3) did not show statistically significant changes, with the exception of chsA and the cell wall stress sensor wscB which were downregulated in celAΔ.

Characterization of the celAΔ strain. (a) Macrophenotype of the celAΔ strain on solid medium. Bar refers to 1 cm. (b) Sensitivity to the cell wall drugs CR and DCB. Numbers refer to spore dilutions. (c) Balloons along the hyphae and aberrant conidiophores in the celAΔ strain. Bars refer to 10 µm.

RT-qPCR analysis on a set of cell wall-related genes in the control and celAΔ strain. The data were normalized using rpl3 and CRP2. Asterisks indicate statistically significant differences (p < 0.05).
Transcriptome of celAΔ
Since a strong phenotype was observed and since the majority of the cell wall biosynthetic genes screened did not show dramatic changes in expression at the RT-qPCR, we decided to perform a transcriptomic analysis to uncover eventual differences in the expression of genes acting in other cellular pathways.
After library preparation, quantification and sequencing, we performed data filtering by setting the log2 FC > 1 in absolute value (see Materials and Methods). A total of 1305 genes showed statistically significant changes (Suppl. Information). The Gene Ontology term Enrichment (GOE) analysis carried out using ClueGO (in Cytoscape) with the genes upregulated in celAΔ (613 in total, see Suppl. Information) revealed an enrichment of functionally-related genes involved in secondary metabolite biosynthesis, such as members of the sterigmatocystin cluster (stcL, stcW, stcV, stcU, stcS, stcK, stcJ, stcF, aflR, stcB, stcA,), as well as genes involved in yellow-polyketide F9775A/F9775B biosynthesis (orsA, orsB, orsC) and monodictyphenone biosynthesis (mdpA, mdpI, mdpH) (Fig. 4; Suppl. Information). Additionally, two putative SM regulatory genes were also responding to the celAΔ deletion by up-regulation (laeA, fhbB). The results obtained with the GO Term Finder at the AspGD confirmed the enrichment of transcripts involved in secondary metabolic processes in celAΔ, with 19 genes involved in toxin biosynthesis (Suppl. Information). The targeted gene expression analysis via RT-qPCR performed on secondary metabolism-related genes (involved in sterigmatocystin, yellow-polyketide F9775A/F9775B and monodictyphenone biosynthesis) confirmed upregulation in celAΔ (Fig. 5). Notably, the analysis with the GO Term Finder highlighted a downregulation of genes involved in organonitrogen compound catabolic process: 20 of the identified genes belong to the cellular amino acid catabolic process (Suppl. Information). These data show that, in a manner analogous to what proven in A. fumigatus 11, an alteration of the cell wall status has an effect on primary metabolism (the primary metabolism is affected with consequent lower abundance of amino acids). Such a phenomenon may be operating in the celA mutant, where a stress response is unleashed as a consequence of celA deletion and therefore a downregulation of genes belonging to the amino acid biosynthetic process is observed.

Gene Ontology Enrichment (GOE) analysis of the genes upregulated in celAΔ. Gene Ontology (GO) terms sharing similar associated genes are connected with a line. The circle sizes represent different p-values (inset).

RT-qPCR analysis on ANIA_08443 and a set of secondary metabolism-related genes in the control and celAΔ strain. The data were normalized using rpl3 and CRP2. Asterisks indicate statistically significant differences (p < 0.05).
High-resolution imaging using scanning electron microscopy (SEM) highlighted a higher number of conidia and conidiophores in celAΔ (Fig. 6). In this respect, it should be noted that in the RNA-Seq dataset four conidiation-related genes with increased expression in the mutant are present (Suppl. Information). These genes are: ANIA_05086 (log2FCcelAΔ/ctrl = 3.246), ANIA_10628 (log2FCcelAΔ/ctrl = 3.236), ANIA_05015 (log2FCcelAΔ/ctrl = 2.509), flbD (log2FCcelAΔ/ctrl = 1.502). Interestingly, flbD is a member of the fluffy genes required to activate brlA and some fluffy mutants are known to display failure to produce sterigmatocystin12. The higher expression of the Myb transcription factor flbD may explain the higher conidiation observed in the mutant, however this is probably not related to the higher sterigmatocystin biosynthesis-related genes, since it was reported that flbD overexpression did not trigger an increase in stc transcripts13. Some secondary metabolites were quantified using LC-MS/MS at different time-points (24 h–72 h–96 h) and the analysis showed an increase in sterigmatocystin, seco-strigmatocystin, austinol and dehydroaustinol in celAΔ which was the strongest at 96 h (the RNA-Seq time-point) (Fig. 7).

Scanning electron microscope pictures of the control (SAA.111) and celAΔ. The deleted strain forms more conidiophores and conidia than the control (the cultures are 4 days old).

Quantification of some secondary metabolites in the control and celAΔ strain.
In the light of the observed results, it is here proposed that the increased expression (and production) of secondary metabolites is linked to the sensing of a stress caused or mimicked by the deletion of the gene celA.
Under laboratory conditions, many fungal SM gene clusters are cryptic and remain dormant based on a chromatin silencing and inactive signaling pathway14. They are instead active when the organism is subjected to specific nutritional conditions and/or environmental constraints15, 16 and enable the colonization of specific natural niches. Manipulations of growth composition can also trigger increased SM biosynthesis, as recently shown with ionic liquids in A. nidulans 17. Therefore, the increased expression in SM biosynthetic genes likely reveals a stress status, probably generated by the altered cell wall in celA∆. It should be noted that the deleted strain shows increased resistance to cell wall drugs (Fig. 2b): this is not unexpected, since in the literature it has for instance been shown that, in A. fumigatus, deletion of dvrA, a transcription factor partaking in cell wall integrity signaling, triggers increased resistance to Nikkomycin Z18. The phenotypes observed, as well as the altered cell wall drug sensitivity, support the hypothesis of a role of celA in a cell wall-related process.
Interestingly, previous studies in the literature on another model fungus, Hypocrea jecorina, have shown that a compromised cell wall has consequences on cellulase production: the mutant Δtmk2 shows increased cellulase production19, 20, a feature most likely triggered to favor the growth of a damaged fungal cell, in a manner analogous to what is observed in the Δtmk3 mutant under solid-state growth19. Cellulases (as well as other secreted enzymes) may help form a protein layer functioning as an osmotic stabilizer for fungal cells with damaged cell walls19. It is thus already reported in the literature that alterations in the cell wall influence complex cellular processes, namely the production and secretion of cellulases.
In our dataset, the second gene showing the highest expression in celAΔ with respect to the isogenic wild type is ANIA_08443, a gene with no known function. This gene shows a log2 FC increase in expression of 8.74 (which means it is expressed ca. 429 times more in celAΔ than the control). The targeted RT-qPCR analysis confirms upregulation of ANIA_08443 in celAΔ (Fig. 5). It should be noted that this gene is adjacent to the celA locus and up-regulation could be a coincidental consequence of marker gene insertion at the place of the celA gene. However, the BLASTP analysis indicates that the corresponding protein has a DUF3431 domain and predicted to possess a transmembrane region (prediction made with TMHMM v2.0 http://www.cbs.dtu.dk/services/TMHMM/ and Phobius http://phobius.sbc.su.se/) and to expose a large portion including the C-terminus to the cell’s exterior (Suppl. Information; residues 27–384 according to TMHMM and 37–384 according to Phobius). Therefore, we cannot exclude that this protein may represent a sensor of the cell wall status. More specifically, this uncharacterized protein may represent a potential candidate involved in cell wall status sensing and signal transduction to the cell’s interior. Its role in signal transduction and activation of SM biosynthetic gene clusters awaits validation via functional studies, nevertheless the results shown pave the way to future studies on celA, as well as on the link between cell wall alterations and SM production in A. nidulans. In this respect we believe important to mention that a DUF3431 protein was shown to be upregulated upon salt stress in the halotolerant fungus A. glaucus 21 and to confer increased stress tolerance when expressed heterologously in Arabidopsis plants.
Future functional studies should provide evidence for an implication of this gene in cell wall status sensing and SM production in A. nidulans.
Materials and Methods
Deletion of celA in A. nidulans
The strain used in this study is SAA.111 (genotype veA1; biA1; ΔargB:: trpC; riboB2; pyroA4; wA3)22. Gene replacement of ANIA_08444 was performed with the double joint-PCR (DJ-PCR)23: the cassette containing the selectable marker argB (ANIA_04409) was inserted to replace the entire locus ANIA_08444. The argB gene was amplified from plasmid pMS1224. Transformation was carried out as described previously22.
The primers used for the DJ-PCR are indicated in Suppl. Information.
PCRs to create the replacement cassette were performed with the Phusion High-Fidelity PCR Master Mix, according to the manufacturer’s instructions. Optimal annealing temperatures were computed with the NEB Tm calculator v1.9.4 (http://tmcalculator.neb.com/#!/).
Deletion was checked via Southern blotting, which provided clear evidence of deletion (Suppl. Figure 1a), however RNA-Seq, due to its high sensitivity, detected very low background expression levels of celA (average 3.8 RPKM). This expression level was however negligible and probably due to contaminating residual heterokaryons; it was not relevant to the further characterization of celA∆, whose phenotype is reminiscent of already characterized cell wall mutants7. Southern blotting (DNA cut with EcoRI and DIG-labelled probe amplified with primers celA Upstream Fwd and celA Downstream Rev), diagnostic PCR on genomic DNA (gDNA) and RT-qPCR were performed as described previously25. Diagnostic PCRs using gDNA were performed with the Q5 Hot-Start High-Fidelity Master Mix, using the primers celA nested Fwd and celA nested Rev (Suppl. Information); the annealing temperature was set at 64 °C with an extension of 3 minutes and a total of 30 amplification cycles on a Veriti Thermal Cycler. The primers of the reference genes rpl37, rpl3, CRP2 and actin used for the RT-qPCR have been previously reported24. The RT-qPCR primers used to amplify the additional reference gene H2B, as well as those relative to the secondary metabolism genes are in Suppl. Information. Optical microscopy and growth test in the presence of cell wall drugs were performed as previously described26.
RT-qPCR, RNA-Seq and bioinformatics
Libraries were prepared from 3 µg of total RNA extracted from the mycelium after 3 days of growth at 37 °C with the SMARTer Stranded RNA-Seq kit (Clontech). The isolation of mRNAs was performed using the Illumina beads and the TruSeq protocol (Illumina), as previously described27. The synthesis of cDNA and shearing were performed with ten ng of mRNA, according to the manufacturer’s instruction. The enrichment step was carried out using 14 cycles of PCR. The libraries were then checked using a 2100 Bioanalyzer (DNA High sensitivity Kit) to estimate the average fragment size. Library quantification was performed using the KAPA library quantification kit (KAPA Biosystems) and a ViiA7 Real-Time PCR System (Life technologies). The pooled libraries (at a concentration of 20 pM) were sequenced on an Illumina MiSeq (MiSeq reagent kit V3, 150 cycles) generating 76 base pairs paired-end reads. Raw sequences have been deposited at the NCBI Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo, under the accession number GSE94110 (link to access the data https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token = qnojukmmnzifxqv&acc = GSE94110).
Raw FASTA files were imported in CLC Genomics Workbench 9.0.1. Sequences were filtered and trimmed according to the following criteria: sequence length > 35 base pairs (bp), sequence quality score < 0.01, no ambiguity in the sequence, trimming using Illumina adaptors, hard trim of 8 bp at the 5′ end and 2 bp at the 3′ end, resulting in a longest length of 64 bp. For each library, the mapping against the A. nidulans FGSC A4 transcriptome (downloaded at the http://www.aspergillusgenome.org/download/sequence/A_nidulans_FGSC_A4/current/) was performed with the following settings: a maximum hit per reads of 3, similarity fraction = 0.9, a length fraction = 0.9, a mismatch, insertion and deletion cost of 3 (stringent mapping). The expression values were then calculated using the RPKM method28. In order to highlight the differentially expressed genes, a t-test with 2 groups (control and celA∆, each composed of three biological replicates) was performed. Only genes with a p-value below 0.05 were selected. A cut-off threshold was also applied to the fold change (log2 FC absolute value > 1).
The GOE analysis was carried out using loci with log2 FC > 1 (i.e. overexpressed in celA∆) using ClueGO (v2.3.2)29 within Cytoscape (v3.4.1) with the following parameters: gene ontology from level 3 to level 8, kappa score set at 0.6, p-value < 0.05, enrichment analysis with a Benjamini-Hochberg correction. The GO analysis was also carried out using the GO Term Finder at the AspGD (http://www.aspergillusgenome.org/cgi-bin/GO/goTermFinder).
Full-length protein sequences (accessions indicated in the tree) were aligned with ClustalOmega (http://www.ebi.ac.uk/Tools/msa/clustalo) and the alignment was submitted to PHYML (http://www.phylogeny.fr). The tree was visualized with iTOL-Interactive Tree Of Life (http://itol.embl.de/).
SEM and metabolite profiling by LC-MS/MS
The analysis at the SEM was carried out as previously described26. LC-MS/MS screening of target fungal metabolites was performed with a QTrap 5500 LC-MS/MS System (Applied Biosystems, Foster City, CA) equipped with a TurboIonSpray electrospray ionization (ESI) source and an 1290 Series HPLC System (Agilent, Waldbronn, Germany). Chromatographic separation was performed at 25 °C on a Gemini® C18-column, 150 × 4.6 mm i.d., 5 µm particle size, equipped with a C18 4 × 3 mm i.d. security guard cartridge (all from Phenomenex, Torrance, CA, US). The chromatographic method, as well as the chromatographic and mass spectrometric parameters, have been previously described30, but the method has in the meantime been expanded to cover 650 metabolites (manuscript in preparation).
References
Horiuchi, H. Functional diversity of chitin synthases of Aspergillus nidulans in hyphal growth, conidiophore development and septum formation. Med. Mycol. 47(Suppl 1), S47–52 (2009).
de Groot, P. W. J. et al. Comprehensive genomic analysis of cell wall genes in Aspergillus nidulans. Fungal Genet. Biol. FG B 46(Suppl 1), S72–81 (2009).
Horiuchi, H., Fujiwara, M., Yamashita, S., Ohta, A. & Takagi, M. Proliferation of intrahyphal hyphae caused by disruption of csmA, which encodes a class V chitin synthase with a myosin motor-like domain in Aspergillus nidulans. J. Bacteriol. 181, 3721–3729 (1999).
Fujiwara, M. et al. Evidence that the Aspergillus nidulans class I and class II chitin synthase genes, chsC and chsA, share critical roles in hyphal wall integrity and conidiophore development. J. Biochem. (Tokyo) 127, 359–366 (2000).
Ichinomiya, M. et al. Repression of chsB expression reveals the functional importance of class IV chitin synthase gene chsD in hyphal growth and conidiation of Aspergillus nidulans. Microbiology 148, 1335–1347 (2002).
Ichinomiya, M., Yamada, E., Yamashita, S., Ohta, A. & Horiuchi, H. Class I and Class II Chitin Synthases Are Involved in Septum Formation in the Filamentous Fungus Aspergillus nidulans. Eukaryot. Cell 4, 1125–1136 (2005).
Takeshita, N., Yamashita, S., Ohta, A. & Horiuchi, H. Aspergillus nidulans class V and VI chitin synthases CsmA and CsmB, each with a myosin motor-like domain, perform compensatory functions that are essential for hyphal tip growth. Mol. Microbiol 59, 1380–1394 (2006).
Yoshimi, A. et al. Functional Analysis of the α-1,3-Glucan Synthase Genes agsA and agsB in Aspergillus nidulans: AgsB Is the Major α-1,3-Glucan Synthase in This Fungus. PLOS ONE 8, e54893 (2013).
Samar, D., Kieler, J. B. & Klutts, J. S. Identification and deletion of Tft1, a predicted glycosyltransferase necessary for cell wall β-1,3;1,4-glucan synthesis in Aspergillus fumigatus. PloS One 10, e0117336 (2015).
García-Rodriguez, L. J. et al. Characterization of the chitin biosynthesis process as a compensatory mechanism in the fks1 mutant of Saccharomyces cerevisiae. FEBS Lett 478, 84–88 (2000).
Jain, R. et al. The MAP kinase MpkA controls cell wall integrity, oxidative stress response, gliotoxin production and iron adaptation in Aspergillus fumigatus. Mol. Microbiol 82, 39–53 (2011).
Wieser, J., Yu, J. H. & Adams, T. H. Dominant mutations affecting both sporulation and sterigmatocystin biosynthesis in Aspergillus nidulans. Curr. Genet. 32, 218–224 (1997).
Hicks, J. K., Yu, J.-H., Keller, N. P. & Adams, T. H. Aspergillus sporulation and mycotoxin production both require inactivation of the FadA Gα protein-dependent signaling pathway. EMBO J 16, 4916–4923 (1997).
Gacek, A. & Strauss, J. The chromatin code of fungal secondary metabolite gene clusters. Appl. Microbiol. Biotechnol. 95, 1389–1404 (2012).
Lim, F. Y., Sanchez, J. F., Wang, C. C. C. & Keller, N. P. Toward Awakening Cryptic Secondary Metabolite Gene Clusters in Filamentous Fungi. Methods Enzymol 517, 303–324 (2012).
Brakhage, A. A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32 (2013).
Alves, P. C. et al. Transcriptomic and metabolomic profiling of ionic liquid stimuli unveils enhanced secondary metabolism in Aspergillus nidulans. BMC Genomics 17, 284 (2016).
Ejzykowicz, D. E. et al. Role of Aspergillus fumigatus DvrA in host cell interactions and virulence. Eukaryot. Cell 9, 1432–1440 (2010).
Wang, M. et al. A Mitogen-Activated Protein Kinase Tmk3 Participates in High Osmolarity Resistance, Cell Wall Integrity Maintenance and Cellulase Production Regulation in Trichoderma reesei. Plos One 8, e72189 (2013).
Wang, M. et al. Identification of the role of a MAP kinase Tmk2 in Hypocrea jecorina (Trichoderma reesei). Sci. Reports 5, 11875 (2014).
Fang, J. et al. Isolation of Salt Stress-Related Genes from Aspergillus glaucus CCHA by Random Overexpression In Escherichia coli. Sci. World J. 2014 (2014).
Bernreiter, A. et al. Nuclear Export of the Transcription Factor NirA Is a Regulatory Checkpoint for Nitrate Induction in Aspergillus nidulans. Mol. Cell. Biol. 27, 791–802 (2007).
Yu, J.-H. et al. Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 41, 973–981 (2004).
Schinko, T., Gallmetzer, A., Amillis, S. & Strauss, J. Pseudo-constitutivity of nitrate-responsive genes in nitrate reductase mutants. Fungal Genet. Biol. 54, 34–41 (2013).
Guerriero, G. et al. A WDR Gene Is a Conserved Member of a Chitin Synthase Gene Cluster and Influences the Cell Wall in Aspergillus nidulans. Int. J. Mol. Sci. 17, 1031 (2016).
Guerriero, G. et al. Sensitivity of Aspergillus nidulans to the cellulose synthase inhibitor dichlobenil: insights from wall-related genes’ expression and ultrastructural hyphal morphologies. PloS One 8, e80038 (2013).
Behr, M. et al. Studying Secondary Growth and Bast Fiber Development: The Hemp Hypocotyl Peeks behind the Wall. Front. Plant Sci. 7, 1733 (2016).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008).
Bindea, G. et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinforma. Oxf. Engl 25, 1091–1093 (2009).
Malachová, A., Sulyok, M., Beltrán, E., Berthiller, F. & Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 1362, 145–156 (2014).
Acknowledgements
GG gratefully acknowledges the support by the Austrian Science Fund (FWF; http://www.fwf.ac.at/en/) Grant No. M1315. Work in the lab of JS was supported by the “EpiMed” NFB grant LS12–009.
Author information
Authors and Affiliations
Contributions
G.G. conceived the idea of writing the paper. G.G. constructed the celA∆ strain, analyzed the phenotype at the optical microscope, performed the Southern blotting, diagnostic PCRs, RT-qPCR, RNA-Seq and bioinformatics. L.S. grew the strains in liquid culture, performed the Southern blotting and contributed to the microscopic analysis. S.L. and F.M. contributed to the bioinformatics. M.S. performed secondary metabolite quantification. G.G., L.S., S.L., F.M., M.S., J.F.H. and J.S. interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession codes: Raw reads were deposited at the NCBI Gene Expression Omnibus (GEO), under accession GSE94110.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guerriero, G., Silvestrini, L., Legay, S. et al. Deletion of the celA gene in Aspergillus nidulans triggers overexpression of secondary metabolite biosynthetic genes. Sci Rep 7, 5978 (2017). https://doi.org/10.1038/s41598-017-05920-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05920-x
This article is cited by
-
Stimulating fungal cell wall integrity by exogenous β-glucanase to improve the production of fungal natural products
Applied Microbiology and Biotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
