
Abstract
Fundamental life history processes of mycorrhizal fungi with inconspicuous fruiting bodies can be difficult to elucidate. In this study we investigated the species identities and life history of the orchid mycorrhizal Tulasnella fungi, which associate with the south eastern Australia orchid genus Chiloglottis. Tulasnella prima was the primary partner and was found to be associated with all 17 Chiloglottis species across a range of >1000 km, and to occur in the two edaphic conditions investigated (soil and sphagnum hammocks). Another Tulasnella species (T. sphagneti) appears to be restricted to moist conditions of alpine sphagnum hammocks. The population genetic structure of the widespread species T. prima, was investigated at 10 simple sequence repeat (SSR) markers and at four cross-amplified SSR loci for T. sphagneti. For both taxa, no sharing of multilocus genotypes was found between sites, but clones were found within sites. Evidence for inbreeding within T. prima was found at 3 of 5 sites. Significant genetic differentiation was found within and between taxa. Significant local positive spatial genetic autocorrelation was detected among non-clonal isolates at the scale of two metres. Overall, the population genetic patterns indicated that in Tulasnella mating occurs by inbreeding and dispersal is typically restricted to short-distances.
Similar content being viewed by others

Introduction
Many terrestrial orchids interact with compatible mycorrhizal fungi throughout their lifetime. Since orchid seed lack an endosperm and consist only of an embryo and a seed coat, this association is obligate during seed germination with the mycorrhizal fungus supplying the critical nutrients for germination1, 2. All orchids continue to rely on fungi for water and nutrients to some extent at adulthood3. Investigating the identity and life history of the fungal partners involved in the symbiosis is critical for improving our understanding of fungal biology and the role they play in orchid distribution and abundance4.
The identification of fungi, especially mycorrhizal fungi has always been challenging because many species lack distinguishing morphological characters. Consequently species identification as well as the fundamental life history processes that structure mycorrhizal populations such as reproductive strategies, dispersal mechanisms, as well as the spatial distribution of clones, individuals and populations are unknown for the vast majority of fungi5. This problem is even more acute for many orchid mycorrhizal fungi (OMF) that mostly belong to the fungal phylum Basidiomycota. Outside of their interaction with orchids, these fungi typically grow saprophytically on dead organic material6, 7 where mycelial growth and sexual stages (basidiomata - fruiting bodies) are inconspicuous corticioid structures. To elucidate the life history processes of fungi, knowledge on the occurrence and rate of fruiting (which can differ considerably among fungi)8 is important but little is known for OMF.
Despite the wide geographic distribution of OMF spanning eastern and Western Australia9, 10, as well as their importance in the recruitment and survival of orchids, nothing is known about their dispersal or mating systems. We predict short dispersal distances for fungi with corticioid fruiting bodies, however mating systems in fungi are complex. Commonly Basidiomycota have two mating type loci (=tetrapolar) or sometimes a single mating type locus only (=bipolar), which control the mating system. The number may vary, but typically fungi in the Basidiomycota have many (up to 1000 s) mating type alleles resulting in many mating types11 where fungi with more mating types are expected to have higher levels of outcrossing. Fungi with tetrapolar mating systems are further expected to have more mating types than those with bipolar mating systems (reviewed in ref. 5). Both these mating systems are found in the Agaricomycetes12 in which the three common orchid mycorrhizal genera (Tulasnella, Serendipita and Ceratobasidium) are included. It is therefore impossible to predict the mating system or even level of inbreeding/outcrossing of these fungal genera without population genetic investigation.
An unusual pattern of orchid-fungal interactions is found in the fungus Tulasnella where just one or two species of fungi is associated with multiple species of Australian terrestrial orchids. In the orchid genera from the tribe Diurideae, subtribe Drakaeinae, the breadth of the association of each orchid genus is restricted to a narrow monophyletic group of lineages of Tulasnella fungi, even for orchids collected over several 100–1000 s of kilometres10, 13, 14. For example, in Chiloglottis (9 spp.) only Tulasnella prima Linde & T.W. May and the recently identified Tulasnella sphagneti Linde & T.W. May are known as symbionts, while across Drakaea and Caleana (7 + 5 spp.) Tulasnella secunda Linde & T.W. May is the sole known symbiont10, 15.
This repeated finding of an Australian orchid genus-wide association each with a narrow phylogenetic mycorrhizal fungal lineage10, 13, 14 raises an important question. Is this broad orchid association true, or merely an artefact of the sampling scale and intensity of the present studies? That is, will the pattern dissolve with expanded sampling for fungi across more orchids within the genera concerned with sampling across a larger geographic range and with the inclusion of samples drawn from diverse edaphic conditions?
The orchid genus, Chiloglottis, provides a unique opportunity to explore these questions more fully. This predominantly eastern Australian genus consists of 24 described species, with a range spanning from subtropical mid north Queensland, through temperate New South Wales and Victoria, to cooler southern Tasmania. Across this range representatives of the genus inhabit a range of plant communities including rainforest margins, forest, woodlands, coastal heath and subalpine areas that span altitudes from sea level up to 1600 m (Atlas of Living Australia website at http://www.ala.org.au; NSW Flora online at http://plantnet.rbgsyd.nsw.gov.au/). Furthermore, several species can be found growing within the same sites in two contrasting edaphic conditions, soil versus sphagnum hammocks.
In this study we considerably expanded the number of Chiloglottis species from which fungal isolates were obtained, with orchid samples drawn from across more than 1000 km, including 17 species, and spanning all the known molecular clades of the genus. In addition, we include an intensive fine-scale analysis (1 m to 30 km) of fungal isolates from three species of Chiloglottis that grow in both soil and sphagnum at high altitudes.
Our study is divided into two parts. First, we applied ITS sequencing to determine the phylogenetic diversity of the fungal isolates associated with Chiloglottis across our broad sample. Second we conducted a fine-scale population genetic analysis at 10 Simple Sequence Repeat loci (SSR) to investigate the habitat association, mating and life history characteristics of fungal isolates from orchid species associated with soil versus sphagnum. Thus, from the fungal perspective, what are the reproductive and life history characteristics of the fungus that allow it to occupy such a large geographic range and diversity of edaphic conditions? Secondly, from the orchid’s perspective, is edaphic diversification associated with diversification in fungal associations? Our findings confirm the tight association with a narrow phylogenetic diversity of mycorrhizal fungi across Chiloglottis orchids, but also confirm a second Tulasnella species that is predominantly associated with sphagnum habitats. Our population genetic analyses provide new insights into the fungal biology of the fungal-orchid interaction.
Results
Tulasnella species diversity and identity
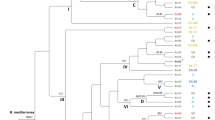
For the ITS phylogenetic analysis, a representative subset of 166 isolates were selected, ensuring coverage of all 17 Chiloglottis species and the 28 sampling sites, spanning >1000 km across south eastern Australia. Two sequences obtained from rhizoids of Sphagnum spp. were also included (Fig. 1). For visual clarity, a large number of identical sequences were omitted from Fig. 1.

Maximum likelihood of ITS sequences from fungal isolates associated with Chiloglottis species. Bayes posterior probability values and bootstrap values are shown. Only posterior probability values >0.70 and bootstrap values >0.80 are shown. For pictorial brevity not all isolates used to compute the posterior probability and bootstrap values were included in the tree. Isolates from the clade representing Tulasnella prima were removed from the tree; these isolates were duplicate or representatives isolates taken from the same site and orchid species shown in the tree. Furthermore, Tulasnella secunda isolates as well as isolates from Arthrochilus oreophilus were removed from the tree for pictorial brevity.
Our analysis found two clades of Tulasnella from Chiloglottis and a clade represented by a single isolate from a Sphagnum rhizoid (clade B) with strong Bayesian Posterior Probabilities (BPP) and bootstrap support (Fig. 1). The largest clade, identified as T. prima since the type of this species is represented in this clade, represented samples drawn from across all 17 Chiloglottis species and from all 28 sites. Furthermore, this Tulasnella species was also found in subalpine regions in orchid samples that grew in both soil (n = 37 plants) and sphagnum hammocks (n = 29 plants). All sequences obtained from direct sequencing of Chiloglottis peloton rich tissue, were represented in this clade (data not shown).
However, subclade A within T. prima is also a highly supported clade with high bootstrap (84%) and Bayes Posterior probability (0.99) values. This clade contained isolates taken from Chiloglottis orchids growing in sphagnum and soil. To test whether subclade A might be a distinct taxon, we investigated percentage sequence divergences. Clade A recorded a mean K2P sequence divergence of 3.55 ± 0.61 to the main T. prima clade (clade containing the type) and thus was at the lower limit of sequence divergence for determining whether the sequences represent a different species. A frequency distribution plot of the percentage sequence diversity among isolates (Supplementary Fig. S1) suggests that 5.5 to 6% sequence divergence would be a conservative cut off to delimit species of Tulasnella associated with Chiloglottis. Similarly, a natural barcode gap across all Tulasnella lineages previously investigated were between 4–6% sequence divergence15, suggesting that clade A is not a distinct species to T. prima.
The clade sister to T. prima was identified as T. sphagneti since it contained the type of this species. Of the isolates sequenced for ITS, all but 2 of the 44 analysed isolates representing T. sphagneti were collected from Chiloglottis orchids growing in sphagnum hammocks. The two exceptions were collected from Chiloglottis growing in soil at the WY site. A sequence obtained from a Sphagnum rhizoid also clustered in the T. sphagneti clade. Another sequence obtained from Sphagnum rhizoids at Barrington Tops formed a highly supported sister node (clade B) to T. sphagneti.
One hundred and thirty ITS haplotypes were observed among the 166 Tulasnella isolates from Chiloglottis. For T. prima, 111 haplotypes were observed from 122 isolates. In T. sphagneti, 26 haplotypes were observed from 44 isolates from Chiloglottis and a sequence from Sphagnum rhizoids. The mean K2P within taxon sequence divergence (converted to percentage) was 1.35% for T. prima and 0.26% for T. sphagneti. The mean sequence divergence between T. prima and T. sphagneti was 8.38% across all sites. Mean sequence divergence for clade B against the main clade of T. prima and T. sphagneti was 8.11% and 7.92%, respectively. Each of the Tulasnella clades from Chiloglottis showed between 13.50 to 19.47% sequences divergence to the outgroup Tulasnella secunda (Table 1).
All 17 Chiloglottis orchid species were present among T. prima isolates (Supplementary Fig. S2) suggesting there is no fungal-Chiloglottis host specificity. T. sphagneti was restricted to two orchid host species; C. turfosa and C. aff. valida, but possibly also C. valida, and only when these hosts were growing in sphagnum hammocks (one exception). A single sample was from Sphagnum rhizoids. Clade B was represented by a single isolate obtained from sphagnum rhizoids, although not from C. sp. (bifaria) orchids which were growing nearby (separated by 2–3 meters only).
Between species population genetic structure of Tulasnella at SSR loci
In the SSR analyses, T. prima (66 MLGs) amplified successfully with all 10 SSR loci while in T. sphagneti (33 MLGs) only four of the 10 loci amplified successfully. The hierarchical AMOVA analysis of the SSR data revealed extensive differentiation between T. prima and T. sphagneti (Table 2), with 44% of the total genetic variance partitioned among the species across the 10 loci (F st = 0.569, six loci set to fixed differences for T. sphagneti). As expected, across the four shared loci, the degree of genetic differentiation between the species was marginally lower (33%) (Table 3). Little allelic sharing occurred among T. prima and T. sphagneti at the four amplifiable SSR loci in common (Supplementary Fig. S3). These two species formed separate clusters in the PCoA with no clustering with respect to orchid species within either Tulasnella species (Fig. 2).

PCoA plot for Tulasnella prima and Tulasnella sphagneti at four SSR loci.
Within population genetic structure of Tulasnella at SSR loci
Within the two Tulasnella species, significant genetic differentiation was detected among populations (6% across the 10 loci, 9% across four loci). A hierarchical multilocus analyses of molecular variance for T. prima from Kosciuszko National Park revealed that most of the variation is found within individuals (59%), with a large portion of the remaining variation explained by variation among individuals (29%) and some by among population differences (13%). However, the reduced number of amplifiable loci for T. sphagneti meant that we could not include this species in all population genetic analyses.
Determination of unique identity of multilocus genotypes
For T. prima from the more intensively sampled area within Kosciuszko National Park, NSW, we genotyped 253 isolates from 99 plants sampled across 5 sites (AC, GST, WY, SH, GG), with an average of two isolates per plant (range 1 to 10 isolates per plant). The conservative PI sibs was estimated to be <0.001 across the 10 SSR loci (Supplementary Fig. S4), indicating strong power to distinguish among different genetic individuals. From our sample of 253 isolates, we detected 67 multilocus genotypes (MLGs), with just seven of the 10 loci required to resolve all 67 MLGs. Therefore, duplicate MLGs were presumed to be clones. Such clones were only found within sites and never shared between sites (Fig. 3), with the most frequently observed clone (27 isolates from 16 plants) at site AC covering an area of over 150 m2. Three MLGs could be isolated from plants growing in either soil or sphagnum at all three sites where T. prima was isolated from plants growing in both soil and sphagnum. We found 13 of the 99 plants had more than one fungal MLG, in all 13 cases the same species of Tulasnella was involved.

Minimum spanning network showing the relationship between individual SSR multilocus genotypes (MLGs) for Tulasnella prima observed in isolates collected in Kosciuszko National Park, NSW from Chiloglottis plants growing in soil or sphagnum hammocks. Each node represents a different MLG. Node sizes and colours correspond to the number of individuals and membership, respectively. The number of individuals per MLG greater than one is provided. Edge thickness and colour are proportional to absolute genetic distances.
Within T. sphagneti, we genotyped 33 isolates across five sites (AC, GST, WY, SH, TTR). Probability of unique identity for the 4 amplifiable loci among unrelated (PI) genotypes was P > 0.045 and for related (PI sibs ) genotypes P > 0.227; suggesting that to identify MLGs, five or more polymorphic loci would be required among unrelated isolates and a greater number for related isolates.
Mating system
Within T. prima from the Kosciuszko National Park, two sites (GG and SH) were in HWE, while three sites showed heterozygote deficiencies. Across all loci and individual sites, there were 22 (of 50) deficits of heterozygotes with significant departures (P < 0.05). After Bonferroni correction, 12 (of 50) loci remained as having a significant heterozygote deficit (Table 4 and Supplementary Table S2). Only two loci from GST showed excess heterozygosity with significantly (P < 0.001) negative F values. Positive and significant (P < 0.001) mean F values were recorded for three sites (AC, GST, WY) showing a deficit of heterozygous individuals compared with expectations under random mating (Table 4), suggesting inbreeding occurs within these sites. When tests of HWE were assessed for the T. prima isolates from Kosciuszko as a single population, we found that all loci significantly (P < 0.001) departed from HWE, as a result of a deficit of heterozygotes (data not shown).
In T. sphagneti, two of the four loci were monomorphic for each site (Tul 2 and Tul 16), and only sites GST and SH showed HWE at the two other amplifiable loci (Tables 5 and S3). After Bonferroni correction, only Tul 12 showed a significant (P < 0.01) heterozygote deficit (Table 5). With all isolates grouped as a single population only one locus (Tul 11) of the three polymorphic loci was in HWE with Tul 12 and Tul 16 significantly (P < 0.001) departing from HWE (data not shown).
Patterns of genetic structure associated with fungal species, geographic origin, edaphic conditions and host identity
Pairwise site F st values ranged from 0.051–0.216 for T. prima and 0.060 to 0.545 for T. sphagneti. All within taxa pairwise site comparisons were significantly different, except those between sites SH and WY (Table 6); these two sites were approximately 7.3 km apart which is at the low end of the geographic distances between sites (5.8 to 26 km). Pairwise F st values within T. sphagneti for site GST were particularly high, which was due to the presence of a private allele at locus Tul 12 that was shared by all GST isolates.
When similar sampling scales were compared for T. prima and T. sphagneti in the Kosciuszko NP region (sites up to 16 km apart), a strong positive and significant relationship was detected between geographic and genetic distance for both species (rxy = 0.225, P ≥ 0.001 and rxy = 0.577, P ≥ 0.001 respectively). Interestingly, across T. prima sampled from the Kosciuszko NP region (sites up to 26 km apart), no individual level isolation by distance was detected (rxy = 0.113, P ≥ 0.065), nor for the Australia wide dataset.
At the three sites where T. prima isolates were sampled from both soil and sphagnum, no significant genetic differentiation was found between the two habitat groups of isolates at two sites (SH- F st 0.068, P ≥ 0.050; AC- F st 0.022, P ≥ 0.188), but significant genetic differentiation was found at site GST (F st 0.133, P ≥ 0.017), which is likely an artefact of the differences in sample size (N = 4 from soil, N = 17 from Sphagnum). Within T. prima, the overlay of geographic origin onto the minimum spanning SSR networks revealed that genotype sharing was geographically restricted (Supplementary Fig. S5). However, there was no clustering associated with orchid species identity (Supplementary Fig. S2). Similarly, no clustering of genotypes for sphagnum versus soil was found within or among sites (Fig. 3).
Isolates which were ITS sequenced and identified as T. sphagneti clustered together in a PCoA with the 15 Tulasnella isolates which were SSR typed only (Fig. 2), suggesting they all belong to T. sphagneti. All but two of the 44 isolates (or 24 of the 26 Chiloglottis plants) representing T. sphagneti were collected from Chiloglottis orchids growing in sphagnum hammocks. The two isolates collected from plants growing in soil came from a single site (WY), however an intensive bushfire five years prior to the collection date may have caused the recession of sphagnum from the area, suggesting a sphagnum habitat specificity for T. sphagneti.
Fine-scale within site genetic structure
At each of the five sites in Kosciuszko National Park, spatial autocorrelation analyses for T. prima isolates showed significant and positive autocorrelation values for the clone uncorrected (range r = 0.142–0.452) or corrected (range r = 0.06–0.353) datasets (Fig. 4). At all five sites the autocorrelation values were greater with clones than without. Fine scale genetic structure at the within-site level with a distance class of two metres showed that r is positive and significant at the shortest distance class of two metres and then generally declines for both the clone uncorrected and corrected datasets at site GST (Fig. 5).

The figure shows the autocorrelation (r) values among five sites located at Kosciuszko National Park for datasets with clones included and clone corrected, respectively. The 95% confidence intervals about the r value were determined via bootstrapping and upper and lower bounds for the 95% confidence intervals about the null hypothesis of no genetic structure (r = 0) determined by random permutation.

Spatial autocorrelation for site GST located at Kosciuszko National Park for datasets with clones included and clone corrected. The 95% confidence intervals about the r value were determined via bootstrapping and upper and lower bounds for the 95% confidence intervals about the null hypothesis of no spatial structure (r = 0) determined by random permutation.
Discussion
Building on our previous work, this study represents the largest and most comprehensive study of orchid-fungal associations across any orchid genus. In total, mycorrhizal fungi from 17 Chiloglottis species representing 28 sampling sites, spanning all four subclades and >1000 km across southeastern Australia, were investigated. Our study also captured a representative number of Chiloglottis orchid species growing in the same area but in different habitats (soil vs sphagnum hammocks), representing intensive sampling at small spatial scales (within site <1 m–15 m; within regions 5 to 10’s of km), as well as regional comparisons (between regions >200 km to over 1000 km).
Mycorrhizal diversity associated with the genus Chiloglottis
Consistent with our previous studies on a limited number of Chiloglottis species10, 14, all 17 Chiloglottis orchid species investigated associated with a dominant Tulasnella species, T. prima. A second and recently discovered Tulasnella species, T. sphagneti 15, was found associated with orchids that grow in subalpine sphagnum hammocks. ITS sequence divergence between T. prima and T. sphagneti (8.38% K2P) fell well above the widely accepted 3% sequence divergence cut-off value for species delimitation16, and the 3–5% divergence proposed for delimiting Tulasnella species17,18,19. Direct sequencing of peloton rich tissues from Chiloglottis plants in this and a previous study14, did not reveal additional orchid mycorrhizal fungal lineages to the cultured isolates, indicating that unculturable fungi are likely to be negligible in this study system. A Ceratobasidium isolate was isolated once and found twice with direct sequencing, however, germination tests with this isolate (data not shown) failed to germinate Chiloglottis seed and hence it is likely a contaminant.
This orchid mycorrhizal association of limited fungal diversity with an entire plant genus, is in stark contrast to the generalist arbuscular mycorrhizae and to a somewhat lesser degree ectomycorrhizae, which all have broad host ranges20. Tulasnella prima has not been found to associate with any other orchid host and thus appears to be restricted to one genus. In general, orchids tend to associate with phylogenetically related groups of fungi. For example, typically an orchid genus associates with several operational taxonomic units (OTUs) of either Tulasnella, Serendipita or Ceratobasidium 21,22,23,24,25,26. Although such associations are interpreted as “specific”, the level of specificity we have discovered in the Tulasnella-Chiloglottis system appears to be unprecedented. In studies of European terrestrial orchids, 15 OTUs from the Tulasnellaceae were associated with four species of Anacamptis. Thirteen of these 15 OTUs were associated with seven Ophrys and two Orchis species and nine OTUs with three Serapias species27. Furthermore, within sites, up to 15 OTUs were co-occurring and 85% of orchid plants associated with more than three different OTUs27. A corresponding result was found along a single 1000 m transect in Italy where 16 Tulasnellaceae OTUs were recovered for 20 species of orchids from the genera Anacamptis, Neotinea, Ophrys, Orchis, and Serapias 28. Similarly, low specificity in another study showed up to seven Tulasnella OTUs may associate with Orchis species within single populations23. The same pattern is found in Andean tropical rainforests where up to 6 Tulasnella OTUs may associate with Stelis orchid species and Pleurothallis lilijae 26, 29. Consistent among these and our studies, is the finding that multiple species of an orchid genus can share the same fungal OTU.
Our findings raise the question of why only a small diversity of Tulasnella fungi associate with an Australian orchid genus spanning a vast geographic range, and across a diversity of habitats. The only other finding of such narrow diversity is found in Australian orchids belonging to the same subtribe (Drakaeinae) as Chiloglottis, ie Drakaea and Caleana (1 OTU in 7 Drakaea and 5 Caleana spp.)10, 13. One exception in this orchid subtribe is found in Arthrochilus oreophilus where 3 OTUs occur in a narrow sample of this subtropical species10. In contrast to the Drakaeinae, five species of Diuris (Tribe Diurideae, subtribe Diuridinae) in Australia associate with several OTUs30.
Currently we have little understanding of why some orchid species interact with a single fungal OTU and other species associate with multiple OTUs. Evolutionary and ecological constraints may account for the patterns of association. Martos et al.31 suggest that where there is asymmetry in associations, phylogenetically constrained heritable traits in the plants may control the suitability of the fungal partner. Furthermore, if related orchids retain much of their ancestral ecological niche, they are likely exposed to the same suite of mycorrhizal fungi31. Some evidence supporting this hypothesis is found in our study, given T. sphagneti was only present in three orchid species that occupy the different edaphic conditions of sphagnum hammocks, whereas all the other species occur in soil and associate with the common and widespread T. prima.
The wide distribution of a common species such as T. prima also may reflect the stable geology of Australia and its associated nutrient poor soils32. By contrast, Europe has had multiple glaciation cycles33 and the soils are comparatively nutrient rich. There is evidence that edaphic conditions and competitiveness influence fungal diversity (see reviews)34, 35; hence in poor soils some fungal species may outcompete less abundant species, whereas in nutrient rich soils sufficient nutrients may sustain a greater diversity of fungi.
Habitat-driven mycorrhizal associations
Whereas T. prima was found in Chiloglottis orchids growing in soil (in Eucalyptus woodlands and forests, subalpine areas) as well as those growing in sphagnum hammocks of subalpine areas, T. sphagneti was strongly associated with Chiloglottis orchids only when they were growing in subalpine sphagnum hammocks. The strong habitat preference of T. sphagneti suggests this fungus is restricted to cool wet environments typical of sphagnum hammocks. Until very recently, the role of habitat as a driver for orchid mycorrhizal differentiation has not been investigated. The first orchid-mycorrhizal study to show habitat preference was on Neottia ovata, which showed that mycorrhizal communities differ substantially between grassland and forest populations of the orchid36. Similarly, habitat was shown to drive different mycorrhizal communities in Epipactis orchids growing in dune slacks vs forests37 and in Dactylorhiza orchids4 in Europe. Our findings reinforce the growing evidence for habitat-driven mycorrhizal associations in orchids and suggest future studies may find this to be a common trend.
Hosts of orchid-mycorrhizae other than the orchid itself are rarely identified. Here we discovered that the three Tulasnella species (T. prima, T. sphagneti and Tulasnella clade B) isolated from orchids also occur in Sphagnum rhizoids (Fig. 1). Aneuraceae thalloid liverworts are known to form associations with Tulasnella in a relationship that may be analogous to mycorrhizas of vascular plants38, 39, however until now associations with Sphagnum have not been investigated. Sphagnum and Bryophytes in general may harbour a vast diversity of Tulasnella that were previously undetected. Interestingly, the three Tulasnella lineages are closely related which may suggest a phylogenetic constraint on host associations and habitat.
Population genetic structure of T. prima
To date, few studies on mycorrhizal fungi and none on orchid-mycorrhizal fungi employed fine-scale spatial analyses of genetic structure to infer the mating system or dispersal. Examining genetic structure at the scale of metres to 100’s of metres allows us to infer the biological processes that lead to positive or negative patterns of genetic distribution.
For Tulasnella, we analysed vegetative material that is dikaryotic (two SSR alleles per locus in an individual) where the two parental haploid nuclei co-exist. Unfortunately only four SSR loci consistently amplified for T. sphagneti, preventing meaningful conclusions regarding its population genetic structure. For T. prima, the SSR markers revealed highly localised genetic structure within sites, along with the trend for heterozygote deficiencies with positive F values reflecting localised inbreeding. Furthermore, three of the tested five populations showed significant deviations of genotype frequencies from Hardy-Weinberg expectations and high positive F values, especially for population GST, consistent with prevalent inbreeding within populations. Only two populations were in HWE, suggesting random mating. Inbreeding is inferred because heterozygote deficiencies were not restricted to certain loci, but was population specific40. However, non-random sampling where homozygotes may have a higher probability of being associated with orchid hosts, or selection against migrants41, 42, as a cause for heterozygote deficiencies40 is probable, but highly speculative in the absence of supporting evidence for such rare phenomena in fungi.
Inbreeding (positive assortative mating) arises when there is mating of related individuals within a population, resulting in individuals being more related than expected under random mating. Although the complex mating systems in basidiomycetes have evolved to promote outcrossing, selfing may occur within a dikaryon that consists of nuclei with compatible mating type alleles. Thus in such cases, in a tetrapolar mating system, 25% of all basidiospores from a single mating event (cluster of basidia) may fuse with a parental haploid individual/genotype (reviewed in ref. 5). Generally, saprotrophic mushrooms have randomly mating populations, but inbreeding is shown increasingly in various mushrooms ranging from the ectomycorrhizal fungus Laccaria amethystina 43, the saprophytic mushrooms Trogia venenata 44 and the bird’s nest fungus Cyathus stercoreus 45. Although further investigation is required to determine whether Tulasnella has a bipolar or tetrapolar mating system, the high levels of inbreeding detected in this study suggest a few mating types only in T. prima.
Significant genetic differentiation (F st = 0.051–0.216) between sites and high spatial autocorrelation found over very short distances (3 m), supports short distance dispersal of basidiospores. Most basidiospores, even among some wind-dispersed fungi, are thought to fall close to the source, which would result in the clustering of genetically similar isolates. Indeed in wind dispersed mushroom species significant positive genetic structure at local scales of 1 to 20 metres46, 47 and up to 400 m48 has been reported. Nonetheless, the signature of spatial clustering among some sites within Kosciuszko suggests that some long distance dispersal of basidiospores does occur, albeit infrequently. It is important to note that estimates of genetic structure produced through F-statistics reflect evolutionary signals of interconnection through gene flow49, whereas spatial autocorrelation provide contemporary genetic signatures. Consequently our results showing restricted short distance dispersal and infrequent long distance dispersal are consistent with studies that have found that most basidiospores tend to fall close to the parent and few are carried long distances via wind dispersal.
Although it is problematic to compare F st values across studies without standardising F st 50, we have found that the level of genetic differentiation between Tulasnella populations in our study were higher at comparable geographic distances than observed in other Basidiomycota fungi population genetic analyses (see review by ref. 5). Basidiomata (fruiting structures) of Tulasnella associated with Chiloglottis have not been observed and it possible that basidiomata of Tulasnella are rarely formed. If this is the case, rare fruiting could severely limit basidiospore production and associated gene flow. Indeed, our population genetic findings suggest that clonal expansion, via vegetative growth or fragmentation of mycelium aided by animal dispersal, seems to be the predominate means of dispersal in the study species.
In conclusion, our new findings are consistent with previous studies confirming that across the genus of Chiloglottis, all orchid species associate with the common and widespread fungus T. prima. However, the species of Tulasnella involved in this orchid-interaction can be influenced by the edaphic conditions in which the orchid grows. Under wetter conditions associated with sphagnum hammocks, a second Tulasnella fungus (T. sphagneti) may co-occur with the generalist T. prima. Our results also provide the first insights into the mating system and likely extent of dispersal within Tulasnella. Despite the wide geographic distribution of T. prima the population genetic results strongly indicate that this species predominantly disperses over short distances and mostly via vegetative (asexual) fragments or predominantly inbred basidiospores.
Methods
Orchid sample collection for mycorrhizal isolation, Tulasnella species diversity and identity
For this first component of the study, Tulasnella mycorrhizal associations were investigated in 17 species of Chiloglottis, representing the three main genetic clades of the genus51: Formicifera clade (C. trapeziformis, C. formicifera); Reflexa clade (C. reflexa, C. trilabra, C. seminuda, C. diphylla); Valida clade (C. turfosa, C. valida, C. cornuta, C. grammata, C. jeanesii, C. pluricallata, C. triceratops, C. gunnii). Two undescribed species were included from the Valida clade, for which there is strong pollinator, chemical and genetic evidence that they are distinct, but morphologically cryptic species: C. aff. valida and C. sp. (bifaria)51,52,53. Chiloglottis palachila was also included as the sole representative of a fourth subclade51, 52.
Orchid plant samples were collected from sites spanning >1000 km across south-eastern Australia. In combination with the earlier sampling efforts of Roche et al.14 and Linde et al.10, our final collection covered a total of 28 sites in New South Wales (NSW), Australian Capital Territory (ACT) and Tasmania (TAS), Australia (Supplementary Table S1).
Orchid sample collection for population structure of Tulasnella at SSR loci
Most of the Chiloglottis species within the Valida clade grow in montane to subalpine regions where they can be observed growing both in moist to wet peaty soils with sphagnum, as well as in soil away from sphagnum dominated habitats. Thus this clade provides a unique opportunity to compare fungal interactions within orchid species that grow under contrasting edaphic conditions, sometimes separated by a few metres only. We therefore sampled three species from this group: C. valida, C. aff. valida and C. turfosa. These species were sampled from both soil and sphagnum hammocks across each of 5 sites (AC, GST, WY, SH, GG) within the Kosciuszko National Park, NSW.
As for the orchid genus generally, flowering in these three species is sparse, with populations typically consisting of multiple colonies each with 100’s to 1000’s of plants, but few if any with flowers. The three species cannot be reliably distinguished by leaf morphology. Furthermore, C. valida and C. aff. valida flowers are morphologically indistinguishable53, but distinct from C. turfosa. Therefore, it was only possible to provisionally identify our samples to species based on floral morphology (when available) and on prior chemical and genetic analysis of tagged colonies from the earlier study of Peakall and Whitehead53.
Depending on the shape of the orchid distribution at a particular site, we used either transects or grids to sample the plants. A single orchid plant located approximately every 1 to 2 metres apart was collected resulting in 30–51 plants collected per Kosciuszko population. Individual plants were mapped for one population (GST) to allow fine-scale analyses of genetic relatedness among fungal isolates.
Fungal isolation
Isolations were conducted within 7 days of the field collection of the plant tissue using a modified version of the protocol of Roche et al.14. We used two types of isolation media to grow mycorrhizal isolates: Fungal Isolation Media (FIM)54 and 3MN + A-Z, which is a Melin-Norkrans medium (low CN MMN)55 modified with 15 g.l−1 agar as well as human vitamin and mineral supplements (Centrum “Balanced Formula”, Wyeth Consumer Healthcare, Baulkham Hills, NSW, Australia) instead of thiamine. One Centrum tablet was dissolved in 100 mL water, filter sterilised, and 10 ml added per litre of 3MN medium, post autoclaving. Peloton-rich tissues (collars) of Chiloglottis were washed several times with sterilised distilled water after which the tissue was macerated in sterilised distilled water to release pelotons which were plated onto agar plates containing antibiotics (FIM + tetracycline 25 mg/mL, and 3MN + A-Z + streptomycin 50 mg/mL). Germinating pelotons were transferred to either FIM or 3MN + A-Z media after 3–10 days. The media choice depended on which medium the pelotons germinated on. After 3–4 weeks all colonies were hyphal-tipped and subcultured to ensure colonies consist of a single genotype. Cultures were stored at 5 °C on FIM or 3MN + A-Z agar slants covered with mineral oil.
To investigate the presence of fungal species that could not be cultured in vitro, a portion of the Chiloglottis peloton-rich tissues (2 × C. aff. valida, 2 × C. turfosa, 2 × C. gunnii, C. valida, and C. triceratops) were stored at −80 °C for DNA extraction and direct sequencing (see below). One plant each of C. aff. valida and C. turfosa were growing in sphagnum hammocks. Rhizoids of Sphagnum cristatum or S. novo-zelandicum were also collected from mountain sphagnum hammocks (from Kosciuszko and Namadgi NP where Chiloglottis plants were collected) to assess Tulasnella presence and identity. As with Chiloglottis peloton-rich tissue, Sphagnum rhizoids were washed several times with sterilised distilled water to minimise the presence of saprophytic fungi on the surface. Voucher specimens of the fungi are stored in the culture collection of CCL laboratory at the Australian National University (ANU; see Supplementary Table S1).
Fungal DNA extraction and analysis
Small agar blocks cut from colony edges of isolates were gently homogenised in 2 ml screw-cap tubes containing sterilised distilled water and glass beads. The blocks were homogenised in a FP120 (Thermo Scientific) homogenizer for 15 sec at 4.0 m/s. Petri dishes containing either FIM or 3MN + A-Z broths were inoculated with the homogenised agar blocks and incubated at room temperature in the dark. Mycelium was harvested, stored at −4 °C and lyophilized prior to DNA extraction. DNA extraction of the lyophilized-mycelium, peloton-rich Chiloglottis tissues and Sphagnum rhizoids were performed using a Qiagen DNeasy Plant Mini Kit or DNeasy 96 Plant Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany).
Fungal ITS sequencing and phylogenetic analysis
In a comprehensive evaluation of eight nuclear and mitochondrial loci, Linde et al.10 showed that within Tulasnella a single locus, ITS (nuclear ribosomal internal transcribed spacer region), revealed congruent species delimitation and phylogenetic outcomes. Therefore, for the phylogenetic analysis in this study we only employed ITS. Details of ITS sequencing, phylogenetic analyses and diversity analyses can be found in the Supplementary Information.
SSR genotyping and population genetic analyses
Ten Simple Sequence Repeat loci (SSR) specifically developed for Tulasnella isolates previously obtained from Chiloglottis orchids were amplified and genotyped as per Ruibal et al.56 for 253 isolates. Isolates where SSR amplification failed for some loci were re-amplified at least once to confirm null-alleles. Except where stated, GenAlEx version 6.50150, 57 was used to conduct the analyses as set out below.
Determination of Unique identity of Multi Locus Genotypes
The power of the SSR loci to uniquely identify individuals (given sample size) was assessed using theoretical predictions of the probability of identity for unrelated (PI) and related individuals (PI sibs ). These analyses provide a theoretical prediction of the average probability that two individuals drawn from the same randomly mating population will by chance have the same multi-locus genotype. PI and PI sibs estimates were calculated using the formula provided in Peakall and Smouse57.
Mating system
To assess the mating system for Tulasnella, Hardy Weinberg Equilibrium (HWE) tests were performed across loci within sites to assess departures from equilibrium. For this and the AMOVA analyses below, identical genotypes were removed from the dataset (hereinafter referred to as clone-corrected data). Heterozygote excess or deficit within each site was assessed with the global HWE test using Genepop version 4.2 (available online at URL: http://kimura.univ-montp2.fr/~rousset/Genepop.htm). P-values were estimated using the Markov chain method (10000 dememorisations, 100 batches, 5000 iterations per batch). To account for multiple comparisons, significance levels of both HWE and F were corrected for using Bonferroni corrections58.
Patterns of genetic structure associated with fungal identity, geographic origin, edaphic conditions and host identity
A hierarchical Analysis of Molecular Variance (AMOVA) was performed on the SSR data set to estimate the degree of genetic differentiation (F ST ) and to partition genetic variation within and among a priori defined Tulasnella groups. These groups included: 1. Putative Tulasnella species predefined based on the outcome of the ITS phylogenetic analysis, 2. Sampling locations (populations) within species, 3. Within species grouped by edaphic conditions (soil versus sphagnum). These same three groupings were overlaid onto a minimum spanning network of the SSR MLGs. The network was constructed and visualized in R59 using the package igraph60 and the kamada.kawai layout option in the R package magrittr to display connections in 2D without overlap of edges in the graph. The network was constructed based on Bruvo’s distance61 which assumes a stepwise mutation model in the calculation of SSR genetic distances among individuals.
Fine-scale within site genetic structure
A multilocus autocorrelation method62,63,64 was used to test for local fine-scale spatial genetic structure. This method allows a test of the null hypothesis of a random distribution of genotypes in space, against the alternative hypothesis of a clustered (non-random) distribution of genotypes. Although the method is generic when analysing multi-allelic and multi-locus genetic data, the generated autocorrelation coefficient r is closely correlated with relatedness65, 66. This fine scale spatial genetic structure analysis was conducted at the within-site level at the site GST.
Mantel tests for isolation by distance were performed following Smouse et al.67, with pairwise geographic and individual genetic distances. Statistical significance of AMOVA and Mantel tests was assessed by 9999 random permutations.
References
Rasmussen, H. N. Recent developments in the study of orchid mycorrhiza. Plant Soil 244, 149–163 (2002).
Warcup, J. H. Mycorrhizas in Orchids of South Australia (eds Bates, R. J. & Weber, J. Z.) 21–26 (Flora and Fauna of South Australia Handbook Committee, 1990).
Dearnaley, J. D. W. Further advances in orchid mychorrhizal research. Mycorrhiza 17, 475–486 (2007).
Jacquemyn, H. et al. Habitat-driven variation in mycorrhizal communities in the terrestrial orchid genus Dactylorhiza. Sci. Rep. 6 (2016).
Douhan, G. W., Vincenot, L., Gryta, H. & Selosse, M. A. Population genetics of ectomycorrhizal fungi: from current knowledge to emerging directions. Fungal Biol. 115, 569–597 (2011).
Greslebin, A. G. & Rajchenberg, M. The genus Tulasnella with a new species in the Patagonian Andes forests of Argentina. Myc. Res. 105, 1149–1151 (2001).
Roberts, P. Rhizoctonia-forming fungi. A taxonomic guide. (Royal Botanic Gardens, Kew, 1999).
Ovaskainen, O. et al. Combining high-throughput sequencing with fruit body surveys reveals contrasting life-history strategies in fungi. The ISME Journal 7, 1696–1709 (2013).
Davis, B. J., Phillips, R. D., Wright, M., Linde, C. C. & Dixon, K. W. Continent-wide distribution in mycorrhizal fungi: implications for the biogeography of specialized orchids. Ann. Bot. 116, 413–421 (2015).
Linde, C. C., Phillips, R. D., Crisp, M. D. & Peakall, R. Congruent species delineation of Tulasnella using multiple loci and methods. New Phytol. 201, 6–12 (2014).
Casselton, L. A. & Kuës, U. The origin of multiple mating types in model mushrooms Coprinopsis cinerea and Schizophyllum commune in Sex in Fungi: Molecular Determination and Evolutionary Implications (eds J. Heitman, W. W. Kornstad, J. W. Taylor, & L. A. Casselton) 283-300 (ASM Press, Washington D.C., 2007).
James, T. Y. et al. Polyporales genomes reveal the genetic architecture underlying tetrapolar and bipolar mating systems. Mycologia 105, 1374–1390 (2013).
Phillips, R. D., Barrett, M. D., Dixon, K. W. & Hopper, S. D. Do mycorrhizal symbioses cause rarity in orchids? J. Ecol. 99, 858–869 (2011).
Roche, S. et al. A narrow group of monophyletic Tulasnella (Tulasnellaceae) symbiont lineages are associated with multiple species of Chiloglottis (Orchidaceae): Implications for orchid diversity. Am. J. Bot. 97, 1313–1327 (2010).
Linde, C. C. et al. New species of Tulasnella associated with terrestrial orchids in Australia. IMA Fungus 8, 27–47 (2017).
Nilsson, R. H., Kristiansson, E., Ryberg, M., Hallenberg, N. & Larsson, K.-H. Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 4, 193–201 (2008).
Cruz, D., Suarez, J. P., Kottke, I. & Piepenbring, M. Cryptic species revealed by molecular phylogenetic analysis of sequences obtained from basidiomata of Tulasnella. Mycologia 106, 708–722 (2014).
Girlanda, M. et al. Photosynthetic mediterranean meadow orchids feature partial mycoheterotrophy and specific mycorrhizal associations. Am. J. Bot. 98, 1148–1163 (2011).
Jacquemyn, H. et al. Analysis of network architecture reveals phylogenetic constraints on mycorrhizal specificity in the genus Orchis (Orchidaceae). New Phytol. 192, 518–528 (2011).
van der Heijden, M. G. A., Martin, F. M., Selosse, M. A. & Sanders, I. R. Mycorrhizal ecology and evolution: the past, the present, and the future. New Phytol. 205, 1406–1423 (2015).
Ogura-Tsujita, Y., Gebauer, G., Hashimoto, T., Umata, H. & Yukawa, T. Evidence for novel and specialized mycorrhizal parasitism: the orchid Gastrodia confusa gains carbon from saprotrophic Mycena. Proc. R. Soc. Lond. Ser. B-Biol. 22, 761–767 (2009).
Shefferson, R. P. et al. Evolution of host breadth in broad interactions: mycorrhizal specificity in East Asian and North American rattlesnake plantains (Goodyera spp.) and their fungal hosts. Mol. Ecol. 19, 3008–3017 (2010).
Jacquemyn, H., Honnay, O., Cammue, B. P. A., Brys, R. & Lievens, B. Low specificity and nested subset structure characterize mycorrhizal associations in five closely related species of the genus Orchis. Mol. Ecol. 19, 4086–4095 (2010).
Jacquemyn, H., Waud, M., Merckx, V. S. F. T., Lievens, B. & Brys, R. Mycorrhizal diversity, seed germination and long-term changes in population size across nine populations of the terrestrial orchid Neottia ovata. Mol. Ecol. 24, 3269–3280 (2015).
McCormick, M. K., Whigham, D. F. & O’Neill, J. Mycorrhizal diversity in photosynthetic terrestrial orchids. New Phytol. 163, 425–438 (2004).
Suarez, J. P. et al. Diverse tulasnelloid fungi form mycorrhizas with epiphytic orchids in an Andean cloud forest. Mycol. Res. 110, 1257–1270 (2006).
Pellegrino, G., Luca, A. & Bellusci, F. Relationships between orchid and fungal biodiversity: Mycorrhizal preferences in Mediterranean orchids. Plant Biosystems 150, 180–189 (2016).
Jacquemyn, H., Brys, R., Waud, M., Busschaert, P. & Lievens, B. Mycorrhizal networks and coexistence in species-rich orchid communities. New Phytol. 206, 1127–1134 (2015).
Kottke, I. et al. Guilds of mycorrhizal fungi and their relation to trees, ericads, orchids and liverworts in a neotropical mountain rain forest. Basic Appl. Ecol. 9, 13–23 (2008).
Smith, Z. F., James, E. A. & McLean, C. B. Mycorrhizal specificity of Diuris fragrantissima (Orchidaceae) and persistence in a reintroduced population. Austral. J. Bot. 58, 97–106 (2010).
Martos, F. et al. The role of epiphytism in architecture and evolutionary constraint within mycorrhizal networks of tropical orchids. Mol. Ecol 21, 5098–5109 (2012).
Blewett, R. S. Shaping a Nation: A Geology of Australia (Geoscience Australia and ANU E press, Canberra, 2012).
Johnson, D. The Geology of Australia (Cambridge University Press, 2009).
Bruns, T. D. Thoughts on the processes that maintain local species-diversity of ectomycorrhizal fungi. Plant and Soil 170, 63–73 (1995).
Kernaghan, G. Mycorrhizal diversity: Cause and effect? Pedobiologia 49, 511–520 (2005).
Oja, J., Kohout, P., Tedersoo, L., Kull, T. & Koljalg, U. Temporal patterns of orchid mycorrhizal fungi in meadows and forests as revealed by 454 pyrosequencing. New Phytol. 205, 1608–1618 (2015).
Jacquemyn, H., Waud, M., Lievens, B. & Brys, R. Differences in mycorrhizal communities between Epipactis palustris, E. helleborine and its presumed sister species E. neerlandica. Ann. Bot. 118, 105–114 (2016).
Bidartondo, M. I. & Duckett, J. G. Conservative ecological and evolutionary patterns in liverwort-fungal symbioses. Proc. R. Soc. Lond. Ser. B-Biol 277, 485–492 (2010).
Preussing, M., Nebel, M., Oberwinkler, F. & Weiss, M. Diverging diversity patterns in the Tulasnella (Basidiomycota, Tulasnellales) mycobionts of Aneura pinguis (Marchantiophyta, Metzgeriales) from Europe and Ecuador. Mycorrhiza 20, 147–159 (2010).
Waples, R. S. Testing for Hardy-Weinberg Proportions: Have We Lost the Plot? J. Hered. 106, 1–19 (2015).
Giraud, T. Selection against migrant pathogens: the immigrant inviability barrier in pathogens. Heredity 97, 316–318 (2006).
Nosil, P., Vines, T. H. & Funk, D. J. Perspective: Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evolution 59, 705–719 (2005).
Roy, M. et al. Evidence from population genetics that the ectomycorrhizal basidiomycete Laccaria amethystina is an actual multihost symbiont. Mol. Ecol. 17, 2825–2838 (2008).
Mi, F. et al. Evidence for inbreeding and genetic differentiation among geographic populations of the saprophytic mushroom Trogia venenata from southwestern China. Plos One 11 (2016).
Malloure, B. D. & James, T. Y. Inbreeding depression in urban environments of the bird’s nest fungus Cyathus stercoreus (Nidulariaceae: Basidiomycota). Heredity 110, 355–362 (2013).
Carriconde, F., Gryta, H., Jargeat, P., Mouhamadou, B. & Gardes, M. High sexual reproduction and limited contemporary dispersal in the ectomycorrhizal fungus Tricholoma scalpturatum: new insights from population genetics and spatial autocorrelation analysis. Mol. Ecol. 17, 4433–4445 (2008).
Wadud, M. A., Nara, K., Lian, C. L., Ishida, T. A. & Hogetsu, T. Genet dynamics and ecological functions of the pioneer ectomycorrhizal fungi Laccaria amethystina and Laccaria laccata in a volcanic desert on Mount Fuji. Mycorrhiza 24, 551–563 (2014).
Dunham, S. M., O’Dell, T. E. & Molina, R. Spatial analysis of within-population microsatellite variability reveals restricted gene flow in the Pacific golden chanterelle (Cantharellus formosus). Mycologia 98, 250–259 (2006).
Bossart, J. L. & Prowell, D. P. Genetic estimates of population structure and gene flow: limitations, lessons and new directions. Trends Ecol. Evol. 13, 202–206 (1998).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28, 2537–2539 (2012).
Peakall, R. et al. Pollinator specificity, floral odour chemistry and the phylogeny of Australian sexually deceptive Chiloglottis orchid: implications for pollinator-driven speciation. New Phytol. 188, 437–450 (2010).
Mant, J. G., Schiestl, F. P., Peakall, R. & Weston, P. H. A phylogenetic study of pollinator conservatism among sexually deceptive orchids. Evolution 56, 888–898 (2002).
Peakall, R. & Whitehead, M. R. Floral odour chemistry defines species boundaries and underpins strong reproductive isolation in sexually deceptive orchids. Ann. Bot. 113, 341–355 (2014).
Clements, M. A. & Ellyard, R. K. The symbiotic germination of Australian terrestrial orchids. Amer. Orchid Soc. Bull. 48, 810–816 (1979).
Wright, M. M., Cross, R., Cousens, R. D., May, T. W. & McLean, C. B. Taxonomic and functional characterisation of fungi from the Sebacina vermifera complex from common and rare orchids in the genus Caladenia. Mycorrhiza 20, 375–390 (2010).
Ruibal, M. P., Peakall, R., Smith, L. M. & Linde, C. C. Phylogenetic and microsatellite markers for Tulasnella (Tulasnellaceae) mycorrhizal fungi associated with Australian orchids. Applications in Plant Sci. 1, 1200394 (2013).
Peakall, R. & Smouse, P. E. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295 (2006).
Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989).
Team RC. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2015).
Csardi, G. & Nepusz, T. The igraph software package for complex network research. InterJournal Complex Systems 1695 (2006).
Bruvo, R., Michiels, N. K., D’Souza, T. G. & Schulenburg, H. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol. Ecol. 13, 2101–2106 (2004).
Peakall, R., Ruibal, M. & Lindenmayer, D. B. Spatial autocorrelation analysis offers new insights into gene flow in the Australian bush rat. Rattus fuscipes. Evolution 57, 1182–1195 (2003).
Smouse, P. E. & Peakall, R. Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82, 561–573 (1999).
Smouse, P. E., Peakall, R. & Gonzales, E. A heterogeneity test for fine-scale genetic structure. Mol. Ecol 17, 3389–3400 (2008).
Banks, S. C., Lindenmayer, D. B., Ward, S. J. & Taylor, A. C. The effects of habitat fragmentation via forestry plantation establishment on spatial genotypic structure in the small marsupial carnivore, Antechinus agilis. Mol. Ecol. 14, 1667–1680 (2005).
Double, M. C., Peakall, R., Beck, N. R. & Cockburn, A. Dispersal, philopatry, and infidelity: Dissecting local genetic structure in superb fairy-wrens (Malurus cyaneus). Evolution 59, 625–635 (2005).
Smouse, P. E., Long, J. C. & Sokal, R. R. Multiple-regression and correlation extensions of the mantel test of matrix correspondence. Syst. Zool. 35, 627–632 (1986).
Acknowledgements
This research was supported by Australian Research Council grants (LP098338 and LP110100408) to RP and CCL and by a Swiss National Found grant (PBNEP3-136536) to YT. We would like to thank G. Triponez-Dayer for help during field work and Christine Hayes for technical assistance.
Author information
Authors and Affiliations
Contributions
C.C.L. designed the research. M.P.R., Y.T. and L.M.S. collected the data and performed the molecular analyses, M.P.R. and C.C.L. analysed the data. M.P.R. wrote the first draft of the manuscript text. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruibal, M.P., Triponez, Y., Smith, L.M. et al. Population structure of an orchid mycorrhizal fungus with genus-wide specificity. Sci Rep 7, 5613 (2017). https://doi.org/10.1038/s41598-017-05855-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05855-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
