
Abstract
Three unusual heterodimeric naphthylisoquinoline alkaloids, named ealapasamines A-C (1–3), were isolated from the leaves of the tropical plant Ancistrocladus ealaensis J. Léonard. These ‘mixed’, constitutionally unsymmetric dimers are the first stereochemically fully assigned cross-coupling products of a 5,8′- and a 7,8′-coupled naphthylisoquinoline linked via C-6′ in both naphthalene portions. So far, only two other West and Central Ancistrocladus species were known to produce dimers with a central 6,6″-axis, yet, in contrast to the ealapasamines, usually consisting of two 5,8′-coupled monomers, like e.g., in michellamine B. The new dimers 1–3 contain six elements of chirality, four stereogenic centers and the two outer axes, while the central biaryl axis is configurationally unstable. The elucidation of the complete stereostructures of the ealapasamines was achieved by the interplay of spectroscopic methods including HRESIMS, 1D and 2D NMR (in particular ROESY measurements), in combination with chemical (oxidative degradation) and chiroptical (electronic circular dichroism) investigations. The ealapasamines A-C display high antiplasmodial activities with excellent half-maximum inhibition concentration values in the low nanomolar range.
Similar content being viewed by others


Introduction
Naphthylisoquinoline alkaloids1 from tropical Ancistrocladaceae and Dioncophyllaceae lianas are the first known tetrahydroisoquinolines of polyketidic origin2. They consist of an isoquinoline and a naphthalene part, usually linked by a rotationally hindered C,C- or N,C-axis. More than 180 such alkaloids have meanwhile been isolated, showing a broad structural diversity1, 3, 4. Most remarkable is the ability of some Ancistrocladus species to produce dimers1, 5,6,7,8,9, thus giving rise to thrilling quateraryls with unique molecular architectures, possessing up to four stereocenters and three biaryl axes. Some of these compounds show significant anti-HIV effects1, 7,8,9, while others exhibit pronounced activities against pathogens causing different tropical diseases like e.g., malaria5, 6, 10.
During the past years, the Cameroonian liana A. korupensis 7, 8 and the Congolese species A. congolensis 9 have attracted particular attention as a rich source of dimeric naphthylisoquinoline alkaloids with a 6′,6″-coupled central biaryl linkage. From these two species, eleven such dimers have so far been discovered, among them michellamine B (4) (Fig. 1), which consists of two 5,8′-coupled molecular halves.

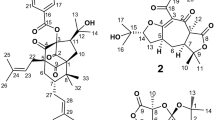
Ealapasamines (A–C) (1–3) from A. ealaensis, and michellamine (B) (4) from A. korupensis.
We herein report on the isolation and structural elucidation of three new, structurally unique heterodimeric naphthylisoquinoline alkaloids, named ealapasamines A (1), B (2), and C (3) (Fig. 1). These secondary metabolites were isolated from the leaves of Ancistrocladus ealaensis Léonard 1, 11, a Central African liana mainly occurring in the Northwestern part of the Democratic Republic of the Congo. The chemical constituents of this plant have so far been studied only scarcely. Earlier work on A. ealaensis 12 has only led to the isolation of the two 5,8′-coupled naphthylisoquinolines ancistroealaines A and B, and of three biosynthetically related naphthoic acid derivatives.
The new compounds 1–3, by contrast, are ‘mixed’, constitutionally unsymmetric dimers consisting of two monomeric parts that are – as for the michellamines – linked via C-6′ of both of the two naphthalene portions. In contrast to all those michellamine-type compounds, however, the ealapasamines A–C are highly unsymmetric, built up from a 5,8′-coupled naphthylisoquinoline and a 7,8′-coupled moiety. As a consequence, they are the first fully assigned heterodimers possessing three different biaryl linkages, and thus show a molecular framework that is substantially different from those of michellamine-type dimers.
The new ealapasamines A–C (1–3) were tested against the pathogens causing malaria tropica, leishmaniasis, Chagas’s disease, and African sleeping sickness, showing remarkably high activities against Plasmodium falciparum.
Results and Discussion
Isolation
LC-MS guided analysis of a crude leaf extract of A. ealaensis revealed the presence of further constituents, with MS profiles typical of dimeric naphthylisoquinoline alkaloids. For the isolation of these compounds, ground leaves were macerated with MeOH, and further partitioned between water and dichloromethane to extract the metabolites. Fractionation of the organic layer by reversed-phase HPLC provided three new dimers.
Structural elucidation of compounds 1–3
Ealapasamine A (1)
The first dimer, with a determined molecular formula of C48H52N2O8 by HRESIMS, was obtained as a colorless solid. The 1H NMR spectrum showed a full set of signals, indicative of an unsymmetric dimer. DEPT-135, HSQC, HMBC, and COSY data (see Supplementary information, SI, Tables 1–4) hinted at the presence of 24 protonated carbon atoms, among them eight aromatic methine groups belonging to six spin systems, two methylene functions, and four aromatic O-methyl groups (Fig. 2). The unsymmetric structure, as also confirmed by the occurrence of 48 signals in the 13C NMR spectrum, excluded that the alkaloid was the known symmetric dimer ancistrogriffithine A13, which has the same molecular formula. Moreover, the presence of the only other unsymmetric dimers known from nature possessing the same molecular formula, mbandakamines A and B6, was easily ruled out since the naphthylisoquinoline alkaloid now discovered in A. ealaensis showed substantially different 1D and 2D NMR spectra, thus evidencing that the isolated dimer was new.

HMBC (red arrows) and ROESY (blue arrows) interactions indicative of the constitutions of the southeastern (A) and the northwestern (B) moieties of 1.
The ‘southeastern’ half of the new dimer displayed a total of four aromatic protons in the 1H NMR spectrum (see Table 1). This portion furthermore showed the presence of one aromatic singlet, H-7′ (δ H 7.28), two aromatic protons with a meta-coupling pattern, H-1′ (δ H 6.80, d, J = 1.18 Hz) and H-3′ (δ H 6.87, d, J = 1.22 Hz), one aromatic methyl group, 2′-Me (δ H,C 2.36, 22.2), one methoxy function, 4′-OMe (δ H,C 4.11, 57.0), and one isolated proton, H-7′ (δ H 6.80, d, J = 1.18 Hz). HMBC correlations were monitored from H-1′ and H-3′ to 2′-Me (Fig. 2A), from H-3′ and 4′-OMe to C-4′ (δ C 158.2), and from H-7′ to C-5′ (C-OH, δ C 152.6), to C-9′ (δ C 136.9), to C-5 (δ C 119.9), and to C-6″ (δ C 120.3). ROESY interactions of H-3′ with both, 2′-Me and 4′-OMe, confirmed the presence of a 6′,8′-substituted naphthalene subunit, which was in agreement with the HMBC, HSQC, COSY, and DEPT-135 data. Moreover, a substituted tetrahydroisoquinoline subportion was assigned according to a shielded aromatic proton at δ H 6.62 (H-7), one aromatic methoxy group at δ H 3.90 (8-OMe), two diastereotopic protons, 4-Heq (δ H 2.64, dd, J = 3.35, 17.78 Hz) and 4-Hax (δ H 2.27, dd, J = 12.14, 17.37 Hz), two methyl groups, 1-Me (δ H 1.76, d, J = 6.70 Hz) and 3-Me (δ H 1.24, d, J = 6.52 Hz), one quartet, H-1 (δ H 4.65, q, J = 6.63 Hz), and one multiplet, H-3 (δ H 3.28, m). The position of H-7 was confirmed by its HMBC cross peaks with C-6 (C-OH, δ C 157.2), C-8 (C-OMe, δ C 158.0), and C-1 (δ C 52.0). HMBC correlations from H-7, H-7′, and 4-Heq to C-5 (δ C 119.9) proved that the two subunits of this monomeric half were 5,8′-coupled and, thus, linked via a rotationally hindered biaryl axis (see Fig. 2A).
The position of the methoxy substituent at C-8 was corroborated by its ROE correlations with H-7 and H-1 (Fig. 2A). The ROESY interactions between H-1 and H-3 supported the relative cis-configuration at the two stereogenic centers in the isoquinoline moiety. The ROESY correlations between 8-OMe and H-1, 1-Me, and H-7, and between 4′-OMe and H-3′, were in agreement with the proposed constitution for this southeastern half (Fig. 3A). The absolute configurations at the chiral centers C-3 and C-3‴ were determined to be R by ruthenium-mediated oxidative degradation14, providing (R)-3-aminobutyric acid. Due to the above-established relative cis-configuration at C-1 versus C-3, the absolute configuration at C-1 was deduced to be S. Based on the ROESY interactions from H-1′ to 4-Hax and 1-Me, and from 4-Heq to H-7′, the axial configuration was assigned to be P in this southeastern half (Fig. 3A).

ROESY interactions defining the relative configurations at the stereogenic centers and axes within the monomeric halves of 1: (A) for the 5,8′-coupled part, and (B) for the 7,8′-linked portion.
The NMR data corresponding to the ‘northwestern’ half of the first dimer displayed one isolated singlet at δ H 7.39 (s, H-7″, δ C 135.2), two doublets with a meta-coupling pattern at δ H 6.88 (d, J = 1.18 Hz, H-1″, δ C 119.8) and 6.86 (d, J = 1.16 Hz, H-3″, δ C 107.9), one methoxy group at δ H 4.11 (s, 4″-OMe, δ C 57.0), a methyl group at δ H 2.35 (s, 2″-Me, δ C 22.1), reminiscent of the naphthalene subunit in the other portion. These assignments were in agreement with the HSQC and HMBC data (Fig. 2B, see also Supplementary Table S2). 1D and 2D NMR data revealed further aromatic and heterocyclic spin systems, namely an aromatic singlet at δ H 6.57 (s, H-5‴, δ C 111.2), a quartet at 4.74 (q, J = 6.77 Hz, H-1‴, δ C 49.9), a multiplet at δ H 3.86 (m, H-3‴, δ C 45.1), two doublets of doublets for the diastereotopic protons at C-4‴ at δ H 3.15 (dd, J = 4.81, 17.58 Hz, 4‴-Heq, δ C 34.5) and 2.88 (dd, J = 11.72, 17.78 Hz, 4‴-Hax, δ C 34.5), one high-field shifted methoxy group at δ H 3.19 (s, 8‴-OMe, δ C 61.0), two methyl groups at δ H 1.63 (d, J = 6.90 Hz, 1‴-Me, δ C 19.5) and 1.51 (d, J = 6.87 Hz, 3‴-Me, δ C 19.3). The HMBC interactions from H-5‴ to C-4‴, to C-7‴ (δ C 121.0), and to C-6‴ (C-OH, δ C 157.6), and from H-1‴ to C-8‴ (δ C 157.5) suggested the presence of a tetrahydroisoquinoline subunit with no substituent at C-5‴ (Table 1). Moreover, the HMBC interactions from H-5‴ and H-7″ to C-7‴, and from H-7″ to C-9″ (δ C 136.4) and C-5″ (δ C 152.5) revealed the naphthalene and isoquinoline subunits of this second molecular half to be connected via a 7‴,8″-biaryl axis (Fig. 3B). This assignment was further proven by the ROESY correlations of 8‴-OMe with H-1‴ and 1‴-Me. In the ROESY spectrum, the cross peaks between H-3‴ and the protons of 1‴-Me established the relative configuration of the stereocenters at C-1”‘ and C-3‴ to be trans (Fig. 3B), in contrast to the observed cis-configured subunit in the southeastern molecular half. The oxidative degradation procedure again delivered only (R)-3-aminobutyric acid, thus the absolute configuration at C-3‴ was attributed to be R. 7,8′-linked naphthylisoquinolines are most challenging to be structurally assigned by NOEs and/or ECD, in particular when being part of a dimer13. A meticulous analysis of the ROEs showed the stereogenic center at C-1‴, with its spin systems H (above the isoquinoline plane) and Me (below), was spatially quite close to the axis, which permitted long-range ROE interactions across the axis over to the naphthalene part. Thus, ROESY correlations between H-7″ and the axial 1‴-Me (both below), and H-1″ with the equatorial H-1‴ (both above) unambiguously established the axis in the 7‴,8″-coupled northwestern half of the dimer to be P-configured.
Since the molecular moieties of this new ‘mixed’, unsymmetric quateraryl were coupled via C-6’ of both naphthalene portions, i.e., in the least-hindered positions, the central biaryl axis was not an additional element of chirality, but can freely rotate. The new dimer thus had the full absolute stereostructure 1, as depicted in Fig. 1. In view of its occurrence in A. ealaensis and according to the Lingala word pasa (=twins), the new dimer 1 was named ealapasamine A.
Prior to this work, only one single alkaloid with a related molecular scaffold had been known, korundamine A (5)15 from the Cameroonian species A. korupensis. This dimer likewise consists of a 5,8′- and a 7,8′-coupled monomer, but its structural elucidation had remained incomplete. The assignment of the relative (and, thus, absolute) axial configuration of the 7,8′-linked molecular half had failed, because no decisive ROE relationships were monitored in its dihydroisoquinoline part (Fig. 4)15. Ealapasamine A (1) is, thus, the very first fully stereochemically elucidated ‘mixed’ heterodimer with two differently coupled naphthylisoquinoline portions that has been fully stereochemically assigned. This assignment was facilitated by the presence of an additional stereocenter at C-1‴, as compared to 5, with its ‘flat’ imino function at C-1‴ and its occurrence in trace amounts only.

Korundamine A (5) previously isolated from A. korupensis 15.
Ealapasamine B (2)
From another dimer-enriched fraction of the leaves of A. ealaensis, a closely related second compound was isolated, albeit in very small quantities only. According to HRESIMS and NMR, this dimer had the same molecular formula as the above- described ealapasamine A (1), and the same constitution, and was, thus a new compound, too (Table 1, see also Supplementary Table S3 and Fig. S5). The relative configuration at C-1‴ versus C-3‴ in the ‘northwestern’ half (Fig. 5A) was deduced to be cis from a ROESY correlation between H-1‴ (δ H 4.69) and H-3‴ (δ H 3.47), while a ROESY interaction between 1-Me (δ H 1.61) and H-3 (δ H 3.70) revealed a relative trans-configuration at C-1 and C-3 in the isoquinoline portion of the ‘southeastern’ half of the molecule (Fig. 5B). In contrast to compound 1, dimer 2 showed a slightly deshielded H-1 (δ H 4.78) and a shielded C-3 (δ C 45.1), and the chemical shifts of H-1‴ and C-3‴ (δ C 51.3), thus likewise corroborating the assignment of the relative configurations in the two tetrahydroisoquinoline portions. The oxidative degradation14 delivered aminobutyric acid as its R-enantiomer only, which implied the absolute configurations of the molecular halves of 2 to be 1 R,3 R in the 5,8′-coupled monomer, and 1 S,3 R in the 7,8′-linked portion. Similar to 1, the ROESY correlations between H-1′ and 4-Hax in the southeastern half (Fig. 5B), and between 1‴-Me and H-1″ for the northwestern half (Fig. 5A) attributed a P-configuration to the two outer biaryl axes. Hence, this new dimer had the full absolute structure 2. It was named ealapasamine B.

ROESY interactions indicative of the relative configurations at the stereogenic centers and axes within the northwestern (A) and southeastern (B) molecular halves of ealapasamine B (2).
Ealapasamine C (3)
A third compound was isolated, along with ealapasamine B, again with a molecular formula identical to those of 1 and 2. Despite some different NMR shifts, its constitution was the same as that of 1 and 2, displaying (see Table 1) slightly shielded quartets at H-1 (δ H 4.65) and H-1‴ (δ H 4.68), and deshielded signals of C-3 (δ C 50.9) and C-3‴ (δ C 51.3). This hinted at relative 1,3-cis-configurations in both tetrahydroisoquinoline moieties, which was further confirmed by ROESY measurements (see Supplementary Fig. S6). Oxidative degradation14 determined the absolute configuration at both, C-3 and C-3‴ to be R, which, in combination with the relative cis-configurations of the two isoquinoline portions, established the stereocenters at C-1 and C-1‴ to be S-configured. Long-range ROESY cross-peaks from H-7′ with 4-Heq, and from H-1″ with 1‴-Me (see Fig. S5), like in the cis-configured moieties of 1 and 2, assigned the two outer biaryl axes to be again P-configured. The new alkaloid had, thus, the structure 3 and was henceforth named ealapasamine C.
Given the freely rotating central biaryl axes, the ECD spectra of 1–3 were all dominated by the chiroptical contributions of the outer axes13. Since these were identically configured for all the three new dimers, their ECD spectra were most similar to each other (Fig. 6A). DFT-based geometric optimizations were also performed for 1-3, leading to the identification of most populated conformers (Fig. 6B, see also Supplementary information).

(A) ECD spectra of ealapasamines (A–C) (1–3) recorded in MeOH. (B) One DFT-optimized conformer of 1.
Biological evaluations
The ealapasamines A–C (1–3) exhibited excellent in vitro antimalarial activities against chloroquine-sensitive (NF54) and chloroquine-resistant (K1) strains of the malaria parasite Plasmodium falciparum (Table 2), with IC50 values of 418 (NF54) and 452 nM (K1) for 1, 210 (NF54) and 138 nM (K1) for 2, and 34 (NF54) and 6.3 nM (K1) for 3. Compound 3 is, thus, the as yet most active naphthylisoquinoline against the resistant strain K1. Its cytotoxicity was comparatively low (6.0 μM), giving a high selectivity index of nearly 1000. Against Trypanosoma b. rhodesiense and Leishmania donovani, by contrast, virtually no activities or very low ones were determined, which demonstrates the high specificity of the antiplasmodial activities of 1–3, making advanced biological evaluations on the most active dimer, 3, a rewarding task.
The ealapasamines are structurally unique in many respects: Among the small subfamily of dimeric naphthylisoquinoline alkaloids (presently ca. 20 compounds)1, 5,6,7,8,9, they are the only fully elucidated ‘mixed’ heterodimers with totally different coupling types at the three biaryl axes (5,8′-, 6′,6″-, and 7‴,8″-coupling). Besides the unprecedented occurrence of such unsymmetric dimers in A. ealaensis (and previously, but not fully assigned, in A. korupensis 15), these alkaloids display exciting antiplasmodial activities.
Experimental Section
General experimental procedures
A Jasco® LC-2000Plus Series System (Gross-Umstadt, Germany) was used for the HPLC-DAD analyses. LC-MS measurements were performed on an Agilent 1100 Series System, equipped with a binary high-pressure mixing pump, with a degasser module, an autosampler, an 1100 series photodiode array (PDA) detector (Agilent Technology, Germany), and an Esquire 3000 Plus ion-trap mass spectrometer with an electrospray ionization interface (Bruker Daltonics, Bremen, Germany). A Bruker Daltonics microTOF spectrometer focus was used for the high-resolution electrospray mass spectrometry. NMR analyses were acquired on AMX 400 and DMX 600 Bruker spectrometers. The offline ECD and ORD spectra were obtained on a Jasco J-715 spectropolarimeter. The data were evaluated using SpecDis_16417. A Shimadzu UV-1800 spectrophotometer was used to perform in triplicate offline UV measurements. The Jasco P-1020-polarimeter operating with a sodium light source (λ = 589 nm) was used for the measurement of the optical rotation. The mechanical shaker operating at the frequency of 160 RPM (rotation per minutes) was from Bottmingen (Switzerland).
Plant material
Leaf material of Ancistrocladus ealaensis 11, 18 was collected in the Botanical Garden of Eala (Mbandaka, Democratic Republic of the Congo), in August 2008 by one of us (V.M.) and in August 2015 by Mr. B.K. Lombe (GPS coordinates 00°03.605 N, 018°18.886E). The material of 2015 was authenticated additionally by LC-DAD-MS to contain the same metabolites as the one of 2008. Voucher specimens are available at the Herbarium Bringmann at the Institute of Organic Chemistry, University of Würzburg (no. 43 and 57).
Extraction and isolation
The air-dried powder of the leaves (600 g) was macerated in MeOH, under mechanical shaking (160 RPM) for 24 h, after filtration the marc was further macerated until exhaustion. The 24h-macerates were mixed after filtration, and evaporated to a viscous solution. The methanolic extract was dissolved in water to permit the precipitation of chlorophyll. The aqueous layer was partitioned first with n-hexane, until the upper phase was cleared of residual chlorophyll, and then exhaustively extracted with CH2Cl2. The organic layer was evaporated to dryness to obtain the metabolites-rich fraction A. Fraction A was then subjected to preparative liquid chromatography using C18-reversed phase silica gel. The mobile system consisted of acetonitrile (MeCN) and ultrapure water containing 0.05% TFA (trifluoroacetic acid). The elution was performed from 0 to 50 per cent H2O in MeCN to afford 100 fractions. Further fractionation of fractions A77 to A88 on five C18-SPE cartridges in series (Sep-Pak C18 Plus Light Cartridge, 130 mg, 55–105 µm) using the same eluting conditions led to several sub-fractions enriched with dimeric-alkaloids which were submitted to semi-preparative HPLC to isolate the dimers.
Semi-preparative HPLC conditions
The isolation from alkaloid-enriched fractions was performed on a SymmetryPrep-C18 column (Waters, 300 × 19 mm, 7 μm) with the mobile phase consisting of A (H2O, 0.05% TFA), B (MeCN, 0.05% TFA), at a flow rate of 10 mL/min. Further purification on a Chromolith SemiPrep RP-18e column (100 × 10 mm) afforded the pure dimeric alkaloids, using a gradient system similar to the one described above on the SymmetryPrep column, but with MeCN replaced by MeOH (C), at the same flow rate.
The fractionation on reversed phase silica gel was guided by LC-MS searching for masses hinting at the presence of dimeric alkaloids. The compounds of interest were found to be distributed unequally between the fractions A77 to A88. For the isolation of 1–3, the fractions of interest were submitted to semi-preparative HPLC on a SymmetryPrep-C18 column using a linear gradient at a flow rate of 10 mL/min: 0–13 min: 10–20% of B, 37 min: 45% of B, 39 min: 50% of B, 42 min: 100% of B, 45 min: 100% of B. Some of the collected peaks (still impure, but containing 1–3) required additional purification steps, which were performed by HPLC on a Chromolith SemiPrep RP-18e column (100 × 10 mm) using a gradient solvent system consisting of A (H2O, 0.05% TFA) and C (MeOH, 0.05% TFA) at a flow rate of 10 mL/min: 0–2 min: 10% of C, 8 min: 30% of C, 11 min: 30% of C, 11.2 min: 35% of C, 16 min: 35% of C, 20 min: 40% of C, 25 min: 45% of C, 27 min: 100% of C, 30 min: 100%, to yield 5 mg of ealapasamine A (1), 1.5 mg of ealapasamine B (2), and 3.5 mg of ealapasamine C (3).
Ealapasamine A (1)
White amorphous powder; \({[{\rm{\alpha }}]}_{{D}}^{23}-21\) (c = 0.09, MeOH); UV (MeOH) λmax (log ε) = 205 (1.32), 217 (0.91), 230 (1.09), 257 (0.54), 262 (0.54), 297 (0.24), 315 (0.28), 322 (0.28), 329 (0.29), 338 (0.28), 344 (0.29) nm; ECD (MeOH, c 0.02) λmax (log ε in cm2 mol−1) 195 (+10.1), 200 (+2.83), 210 (‒7.81), 221 (‒1.71), 227 (‒3.51), 234 (−3.34), 240 (−3.32), 250 (+1.36), 267 (−11.01), 290 (−0.09), 303 (−2.96), 351 (+1.1), 395 (−0.6) nm; ORD (MeOH, c 0.02) λmax (log ε in cm2 mol−1) 200 (+5.3), 204 (+9.0), 207 (+7.7), 219 (−0.4), 226 (+1.8), 238 (+0.6), 244 (−1.4), 259 (+6.7), 279 (−6.6), 298 (−1.4), 317 (−4.7), 348 (−1.2), 363.8 (0.0), 380 (−0.5), 404 (−1.0) nm; 1H NMR and 13C NMR data: see Table 1; HRESIMS m/z 785.37804 [M + H]+ (calcd for C48H53N2O8, 785.37964).
Ealapasamine B (2)
White amorphous powder; \({[{\rm{\alpha }}]}_{D}^{23}-10\) (c 0.04, MeOH); UV (MeOH) λmax (log ε) 205 (1.2), 219 (0.81), 228 (0.85), 257 (0.42), 267 (0.45), 305 (0.16), 312 (0.17), 323 (0.16), 330 (0.16), 338 (0.16), 344 (0.16) nm; ECD (MeOH, c 0.005) λmax (log ε in cm2 mol−1) 195 (+1.3), 200 (−1.26), 207 (−7.68), 236 (+3.57), 240 (+2.43), 267 (−3.5), 290 (+0.7), 304 (−0.94), 334 (−0.23), 386 (+0.17) nm; ORD (MeOH, c 0.005) λmax (log ε in cm2 mol−1) 200 (+4.1), 202 (+5.0), 212 (−4.4), 221 (−2.8), 228 (−3.3), 242 (+1.8), 246 (+1.4), 258 (+2.6), 280 (−2.6), 297 (+0.1), 315 (−1.3), 355 (−0.6), 365 (−0.7), 400 (−0.3) nm; 1H NMR and 13C NMR data: see Table 1; HRESIMS m/z 785.38042 [M + H]+ (calcd for C48H53N2O8, 785.37964).
Ealapasamine C (3)
White amorphous powder; \({[{\rm{\alpha }}]}_{D}^{23}-26\) (c 0.1, MeOH); UV (MeOH) λmax (log ε) 217 (0.83), 229 (0.93), 257 (0.37), 263 (0.4), 300 (0.14), 315 (0.16), 322 (0.15), 330 (0.16), 338 (0.16), 344 (0.17) nm; ECD (MeOH, c 0.01) λmax (log ε in cm2 mol−1) 195 (+7.34), 200 (−1.66), 202 (+5.2), 212 (−11.1), 224 (−6.23), 249 (+1.95), 267 (−13.66), 292 (+0.65), 305 (−1.89), 325 (+1.1), 349 (−0.56), 385 (−0.17) nm; ORD (MeOH, c 0.01) λmax (log ε in cm2 mol−1) 200 (−0.9), 206 (+13.2), 218 (−2.6), 222 (+0.1), 231 (−4.4), 242 (+0.0), 259 (+8.4), 280 (−8.5), 299 (−2.3), 315 (−4.2), 343 (−1.0), 354 (−1.7), 400 (−1.1) nm; 1H NMR and 13C NMR data: see Table 1; HRESIMS m/z 785.37932 [M + H]+ (calcd for C48H53N2O8, 785.37964).
Oxidative degradation
The ruthenium (VIII)-mediated periodate degradation of the ealapasamines A–C (1–3), the Mosher-type derivatization of the resulting amino acids using MeOH/HCl and R-α-methoxy-α-trifluoromethylacetyl chloride (R-MTPA-Cl, prepared from S-MTPA), and the subsequent GC-MSD analysis were carried out as described earlier14.
Computational analysis
The DFT structural geometry optimizations of ealapasamines A-C were calculated using the B3LYP-D3/def2-TZVP method, with ORCA19,20,21.
Biological evaluation
Antiprotozoal in vitro tests were performed on the NF54 (chloroquine-sensitive) and K1 (chloroquine- and pyrimethamine-resistant) strains of Plasmodium falciparum, STIB 900 strain of Trypanosoma brucei rhodesiense (trypomastigotes), Tulahuen C4 strain of Trypanosoma cruzi (amastigotes), and MHOM-ET-67/L82 strain Leishmania donovani (amastigotes). The cytotoxicity on mammalian host cells (rat skeletal myoblast L6 cells) were determined according to established protocols22.
References
Bringmann, G. & Pokorny, F. The naphthylisoquinoline alkaloids in The Alkaloids (ed. Cordell, G. A.) Academic Press: New York, Vol. 46, pp 127–271 (1995).
Bringmann, G., Wohlfarth, M., Rischer, H., Grüne, M. & Schlauer, J. A new biosynthetic pathway to alkaloids in plants: acetogenic isoquinolines. Angew. Chem. Int. Ed. 39, 1464–1466 (2000).
Bringmann, G., Günther, C., Ochse, M., Schupp, O. & Tasler, S. In Progress in the Chemistry of Organic Natural Products (eds Herz, W., Falk, H., Kirby, G. W. & Moore, R. E.) Springer: Wien, New York, Vol. 82, pp 111–123 (2001).
Ibrahim, S. R. M. & Mohamed, G. A. Naphthylisoquinoline alkaloids potential drug leads. Fitoterapia 106, 194–225 (2015).
Xu, M. et al. Shuangancistrotectorines A-E, dimeric naphthylisoquinoline alkaloids with three chiral biaryl axes from the Chinese plant Ancistrocladus tectorius. Chem. Eur. J 16, 4206–4216 (2010).
Bringmann, G. et al. Mbandakamines A and B, unsymmetrically coupled dimeric naphthylisoquinoline alkaloids, from a Congolese Ancistrocladus species. Org. Lett. 15, 2590–2593 (2013).
Boyd, M. R. et al. Anti-HIV Michellamines from Ancistrocladus korupensis. J. Med. Chem. 37, 1740–1745 (1994).
Hallock, Y. F. et al. Michellamines D–F, new HIV-inhibitory dimeric naphthylisoquinoline alkaloids, and korupensamine E, a new antimalarial monomer, from Ancistrocladus korupensis. J. Nat. Prod. 60, 677–683 (1997).
Bringmann, G. et al. HIV-inhibitory michellamine-type dimeric naphthylisoquinoline alkaloids from the Central African liana Ancistrocladus congolensis. Phytochemistry 128, 71–81 (2016).
Zofou, D., Ntie-Kang, F., Sippl, W. & Efange, S. M. N. Bioactive natural products derived from the Central African flora against neglected tropical diseases and HIV. Nat. Prod. Rep. 30, 1098–1120 (2013).
Taylor, C. M., Gereau, R. E. & Walters, G. M. Revision of Ancistrocladus Wall. (Ancistrocladaceae). Ann. Missouri Bot. Gard. 92, 360–399 (2005).
Bringmann, G. et al. Ancistroealaines A and B, two new bioactive naphthylisoquinolines, and related naphthoic acids from Ancistrocladus ealaensis. J. Nat. Prod. 63, 1465–1470 (2000).
Bringmann, G. et al. A photometric screening method for dimeric naphthylisoquinoline alkaloids and complete on-line structural elucidation of a dimer in crude plant extracts, by the LC-MS/LC-NMR/LC-CD triad. Anal. Chem. 73, 2571–2577 (2001).
Bringmann, G., God, R. & Schäffer, M. An improved degradation procedure for determination of the absolute configuration in chiral isoquinoline and β-carboline derivatives. Phytochemistry 43, 1393–1403 (1996).
Hallock, Y. F. et al. Korundamine A, a novel HIV-inhibitory and antimalarial “hybrid” naphthylisoquinoline alkaloid heterodimer from Ancistrocladus korupensis. Bioorg. Med. Chem. 8, 1729–1734 (1998).
Bringmann, G. et al. Jozimine A2: The first dimeric Dioncophyllaceae-type naphthylisoquinoline alkaloid, with three chiral axes and high antiplasmodial activity. Chem. Eur. J. 19, 916–923 (2013).
Bruhn, T., Schaumlöffel, A., Hemberger, Y. & Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 25, 243–249 (2013).
Léonard, J. Une nouvelle et curieuse famille pour la flore phanérogamique du Congo Belge: les Ancistrocladaceae. Bull. Soc. Roy. Bot. Belg. 82, 27–40 (1949).
Neese, F. The ORCA program system. WIREs: Comput. Mol. Sci. 2(2), 73–78 (2012).
ORCA, Version 3.0.3. https://orcaforum.cec.mpg.de/ (2014).
Hemberger, Y., Zhang, G., Brun, R., Kaiser, M. & Bringmann, G. Highly antiplasmodial non-natural oxidative products of dioncophylline A: synthesis, absolute configuration, and conformational stability. Chem. Eur. J. 21, 14507–14518 (2015).
Orhan, I., Sener, B., Kaiser, M., Brun, R. & Tasdemir, D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs 8, 47–58 (2010).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 630, “Agents against Infectious Diseases”, project A2) and by the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg (grant to D.T.T.). Generous support from the Else-Kröner-Fresenius-Stiftung is also acknowledged. The authors thank Mr. B.K. Lombe for the collection of additional leaf material of A. ealaensis in the Eala Park (Province Équateur, DR Congo). Further thank is due to Dr. M. Grüne and Mrs. Altenberger for the NMR measurements, to Dr. M. Büchner and Mrs. J. Adelmann for the HRESIMS analyses, and to Mrs. M. Michel for the degradation experiments. We dedicate this paper to Professor Dr. Dr. h.c. Alfred Forchel, President of the University of Wuerzburg, on the occassion of his 65th birthday.
Author information
Authors and Affiliations
Contributions
G.B. and D.F. designed this project. D.T.T. performed the isolation and structural elucidation experiments. G.B., D.T.T., and D.F. jointly wrote the manuscript. V.M. collected and validated the plant material used for the isolation of the ealapasamines. M.K. and R.B. tested the compounds for their antiparasitic activities and evaluated the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tshitenge, D.T., Feineis, D., Mudogo, V. et al. Antiplasmodial Ealapasamines A-C,‘Mixed’ Naphthylisoquinoline Dimers from the Central African Liana Ancistrocladus ealaensis . Sci Rep 7, 5767 (2017). https://doi.org/10.1038/s41598-017-05719-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05719-w
This article is cited by
-
Naphthylisoquinoline alkaloids, a new structural template inhibitor of Nav1.7 sodium channel
Acta Pharmacologica Sinica (2023)
-
Naturally Occurring Alkaloids, Derivatives, and Semi-synthetic Modifications as Lead Compounds for the Development of New Anti-Trypanosoma cruzi Agents
Current Clinical Microbiology Reports (2021)
-
Antiplasmodial natural products: an update
Malaria Journal (2019)
-
An Unusually Broad Series of Seven Cyclombandakamines, Bridged Dimeric Naphthylisoquinoline Alkaloids from the Congolese Liana Ancistrocladus ealaensis
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
