
Abstract
The origin and evolution of land plants was an important event in the history of life and initiated the establishment of modern terrestrial ecosystems. From water to terrestrial environments, plants needed to overcome the enhanced ultraviolet (UV) radiation and many other DNA-damaging agents. Evolving new genes with the function of DNA repair is critical for the origin and radiation of land plants. In bacteria, the DNA-3-methyladenine glycosylase (MAG) recognizes of a variety of base lesions and initiates the process of the base excision repair for damaged DNA. The homologs of MAG gene are present in all major lineages of streptophytes, and both the phylogenic and sequence similarity analyses revealed that green plant MAG gene originated through an ancient horizontal gene transfer (HGT) event from bacteria. Experimental evidence demonstrated that the expression of the maize ZmMAG gene was induced by UV and zeocin, both of which are known as DNA-damaging agents. Further investigation revealed that Streptophyta MAG genes had undergone positive selection during the initial evolutionary period in the ancestor of land plants. Our findings demonstrated that the ancient HGT of MAG to the ancestor of land plants probably played an important role in preadaptation to DNA-damaging agents in terrestrial environments.
Similar content being viewed by others

Introduction
The origin of land plants (Embryophytes) is one of the most important events in the evolution of life on earth and was the key step in the process of developing modern terrestrial ecosystems. Numerous lines of evidence have revealed that land plants evolved from water-based green algae1,2,3. From water to terrestrial environments, plants have enjoyed the advantages of the land, such as brighter sunlight, more carbon dioxide in the atmosphere and more plentiful mineral nutrients in the soil. On the other hand, land plants have had to overcome challenges in terrestrial environments, including less water in land and no support against gravity. Some new structures, including cuticles, vascular tissue, roots and leaves, have evolved to adapt these abominable environments on land4, 5. Due to their sessile nature, land plants are continuously exposed to DNA-damaging agents, including UVB, ozone, desiccation, rehydration, salinity, low and high temperature, and air and soil pollutants including metals-metalloids. In addition, some other environmental stressors, including chemical mutagens, ionizing radiations, alkylating agents, aromatic compounds and microbial toxins, can also damage the DNA in land plants6, 7. Plants use light energy from the sun for photosynthesis to make their chemicals. However, sunlight is the greatest source of UV radiations. The photosynthetic characteristics and sessile nature of land plants make it unpreventable to avoid UV radiation. As a part of ultraviolet radiation, UVB (280–315 nm) is directly absorbed by DNA and induces various DNA lesions such as cyclobutane pyrimidine dimers (CPDs), 6-4 pyrimidine-pyrimidone photoproducts (6-4 PPs), and other minor types of DNA damage. Every pyrimidine dimer blocks transcription and replication and is sufficient to completely eliminate expression of a transcriptional unit6, 8,9,10. Therefore, effective mechanisms of DNA repair are essential for adaptation to terrestrial environments, and hence to ensure the stability of the genome for plants11.
DNA glycosylases catalyze the first step of base excision repair (BER) pathway, the mechanism by which damaged bases in DNA are removed and replaced12. These enzymes remove the damaged nitrogenous base while leaving the sugar-phosphate backbone intact, creating an apurinic/apyrimidinic site (AP site)13, 14. DNA-3-methyladenine glycosylase (MAG), which is also known as 3-alkyladenine DNA glycosylase (AAG) or N-methylpurine DNA glycosylase (MPG), is thought to be involved in the recognition of a variety of base lesions, including alkylated and deaminated purines and to initiate their repair via the BER pathway15. The broad substrate recognition by MAG serves to provide resistance to a wide variety of DNA damaging agents16. Although nearly all MAGs share the ability to rescue 3-methyladenine DNA glycosylase-deficient Escherichia coli from death induced by the alkylating agent methylmethane sulfonate (MMS), the MAGs can be divided into several subfamilies that act on a wide variety of damaged DNA bases16. However, some MAGs were found to remove normal bases from the genome, suggesting that they may have detrimental consequences under certain conditions17,18,19.
In land plants, the gene encoding MAG was first isolated in Arabidopsis thaliana, and this gene complements the MMS-sensitive phenotype of an E. coli double mutant deficient in 3-methyladenine glycosylases20, 21. The MAG genes have also been detected in some other higher plants, including maize22, 23, wheat24, grape25 and Brachypodium distachyon 26. These results suggested that the homologs of the Arabidopsis MAG gene might be present in a wide range of land plants, and the ubiquity of this gene in land plants suggests that its functions include a wide range of selectivity. Here, we provide information that helps answer some questions regarding the evolution of land plant MAG gene by performing an extensive search of its homologs in current sequence databases and by analyzing their phylogeny.
Results
MAG genes are widely present in streptophytes
Comprehensive BLAST searches revealed that MAG genes are widely present in various streptophytes, from charophyte algae to bryophytes, pteridophytes and seed plants (Supplementary Table S1). However, no MAG homologs were detected in any genomes of chlorophyte algae. In the genomes of the moss Physcomitrella patens and the gymnosperm Picea abies that have been fully sequenced, no MAG homologs were found. In addition, no EST hits of MAG genes were observed in these two species. However, we noticed that another moss genome, the Sphagnum fallax, contained one MAG gene. Some EST hits in gymnosperm P. glauca were matched to the MAG homologs. These results suggested that the moss P. patens and the gymnosperm P. abies have lost the MAG gene during evolution. In addition to the genome of Klebsormidium flaccidum, another charophyte alga, Penium margaritaceum, possesses ESTs which show high similarity with the MAG genes, suggesting that this gene might be present widely in charophyte algae.
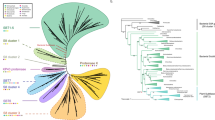
To investigate the evolution of MAG genes in green plants, we examined genomes representing the main lineages of streptophytes, including the charophyte alga K. flaccidum, the moss S. fallax, the liverwort Marchantia polymorpha, the lycophyte Selaginella moellendorffii, the gymnosperm P. glauca, the basal angiosperm Amborella trichopoda, and eight monocot and 36 dicot angiosperms (Supplementary Table S1 and Fig. 1). All these genomes contain only one MAG gene except for the dicot Camelina sativa, which possesses three MAG genes. In the phylogeny, all C. sativa MAG paralogs were located at the termini of branches, suggesting that these paralogs were formed through recent duplication events after split with other species. Further evidence revealed that these three paralogous genes resulted from segmental duplication because they are located in different chromosomes and there were highly conserved genes within the flanking regions of these paralogous MAG genes. Nearly all of the selected MAG genes contained five introns in their coding regions, and their positions and phases showed characteristics of conservation. This investigation suggested the plant MAG genes have highly similar gene structure and the main features of the MAG gene was established before the origin of land plants. However, we did notice that two of the plant MAG genes showed different exon/intron structure than others, the moss S. fallax gene SfMAG and the liverwort M. polymorpha gene MpMAG. Through comparing its sequence with others, we conclude that the SfMAG gene had acquired 2 introns in its 5′ region. The gene MpMAG only contained one intron in coding region, and its position and phase of this gene is different from other plant MAG genes, suggesting that it might be formed through reconstruction of exon/intron structure (Fig. 1).

Phylogenetic tree of green plant DNA-3-methyladenine glycosylase (MAG) genes and their exon/intron structures. The numbers above the branches represent the bootstrap values for the maximum likelihood and distance analyses, respectively. The asterisks indicate values <50%. The exons are indicated by boxes, and introns are indicated by lines. The number above an intron indicates the phase.
Streptophyta MAG gene was acquired through an ancient horizontal gene transfer event
Similarity searches also revealed that homologs of Streptophyta MAG genes are widely present in many cellular organisms, including red algae, fungi, ciliates, trichomonads, apicomplexans, metazoan, bacteria and Archaea. The universality of their distribution in bacteria also suggested that this gene first emerged in bacteria. In addition, we noticed that the green plant MAG proteins showed higher similarity with homologs in bacteria than those in other eukaryotes. To determine the origin of the Streptophyta MAG genes, we selected representative homologs from each taxonomic group of cellular organisms in the nr database to build a phylogenetic tree (Fig. 2). Each of the proteins selected for phylogenetic analysis possessed a Pur_DNA_glyco domain (PF02245). In the phylogenetic tree, all of the Streptophyta MAG formed a single clade with high bootstrap supports. The monophyly of the Streptophyta MAG genes strongly suggests that they have a single origin and are derived from a unique gene that was present in the ancestor of the Streptophyta. Although some other eukaryotic genomes contained the genes encoding MAGs, they did not follow the same clade with those in green plants, suggesting that the Streptophyta MAG genes have a different origin pattern than those in other eukaryotes. In addition, we noticed that the Streptophyta MAG genes fell within the branch of bacterial genes showing high bootstrap support values in both maximum likelihood and distance analyses. The bacterial genes in this branch come from species of gammaproteobacteria, deltaproteobacteria, and acidobacteria. Furthermore, we observed that the Streptophyta MAG proteins showed the highest similarity to those of bacteria in this clade; in particular, at least three motifs in the C-terminus are only conserved in green plant and these bacterial MAGs (Supplementary Fig. 1). These findings illustrate that the ancestor of streptophytes had possibly acquired the MAG gene through a single ancient HGT event from bacteria prior to the origin of land plants.

Phylogenetic analyses of MAG proteins. The numbers above the branches are the bootstrap values for maximum likelihood and distance analyses. Asterisks indicate values lower than 50%. Brown yellow shading indicates lineages in which land plant MAG genes evolved. Green, red, blue and black branches indicate genes from streptophyta, eukaryota, archaea and bacteria, respectively. All sequences were obtained from NCBI, except for those in streptophyta, and the locus number in NCBI and genus is given for each protein.
Positive selection facilitated the evolution of the MAG genes in streptophytes
To test the selective constraints on the evolution of MAG genes, we used the site-specific model to detect positively selected sites for the sampled Streptophyta MAG genes and six bacterial genes following the same branch as plants. The maximum likelihood estimates of the d N /d s values under M0 model for plant and bacterial groups were 0.0740 and 0.0486 (Supplementary Table S2), respectively. These two estimates are close to zero, suggesting that purifying selection was the predominant force for the evolution of the MAG genes both in streptophytes and selected bacterial lineages. However, the log-likelihood differences between models M3 and M0 were statistically significant for both lineages, indicating that the selective constraint level varied across amino acid positions. To determine whether positive selection promoted the divergence of the MAG genes in both lineages, two likelihood ratio tests were employed to compare the data fit to models M2a vs M1a and M8 vs M7. None of the LRTs were statistically significant, and no amino acid site was found to be influenced by positive selection during the evolution of MAG genes either in streptophytes or in bacteria (Supplementary Table S2). These results suggested that purifying selection was the predominant force in the evolution of both the Streptophyta and bacterial MAG genes, and no positive selection signature was found during their evolution either in these two lineages.
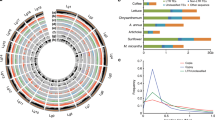
To detect whether streptophytes are characterized by a different pattern of molecular evolution for the MAG genes compared with that in bacteria, an improved branch-site model27 was used to identify the positively selected sites in Streptophyta genes. A new phylogenetic tree was reconstructed using the green plant and bacterial MAG proteins, and the branch of Streptophyta MAG genes was classified as the foreground branch, while those in bacteria as the background branches. In this analysis, only the bacterial genes following to the same branch of plants were selected. We found that the model that permitted a class of positively selected codons with d N /d s > 1 for the Streptophyta branch had a significantly better fit to the data than the model in which this class of codon was restricted to d N /d s = 1 (Table 1). The method of Bayes empirical Bayes28 was further employed to determine the positively selected sites and their posterior probabilities, and 6 codons were found to have a 95% posterior probability of positive selection (Supplementary Fig. 2). The predicted three-dimensional structure of the maize ZmMAG protein possessed the thirteen β-strands (β1-β13) and six regular α-helices (α1-α6). We noticed that only one positively selected sites (V238) is located in the region of helix, while all the other five sites are located in regions connecting the α-helices and β-strands (Fig. 3). These observations suggest that positive selection pressure on these residues might have changed the protein structure, thus accelerated functional divergence. The most reasonable explanation for these results is that the MAG gene underwent adaptive evolution in the streptophyte ancestor during a short period of time after the HGT event, although the dominant force for the evolution in streptophytes of these codons is purifying selection.

Putative positive selection sites of ZmMAG. (A) Secondary structure of ZmMAG. The positive selection residues are indicated with asterisk. (B) 3D structure of ZmMAG. The positive selection residues are marked and are colored in different colors. The structure was predicted using the tool of SWISS-MODEL and was displayed by the software Geneious.
Expression of the maize MAG gene is regulated by DNA-damaging agents
The gene expression pattern can provide valuable information on the function of a protein. In this study, we examined the expression pattern of the maize (Zea mays L.) MAG gene in response to DNA-damaging agents. First, we detected the expression level of this gene following treatment with UV light (Fig. 4). The ZmMAG transcripts gradually increased in the maize seedlings within the first 3 h after exposure to UV treatment, and peaked with the highest expression level at 12 h. Second, we noticed that the treatment of zeocin also up-regulated the expression of ZmMAG gene, and the level of expression reached the highest level at 6 h. However, the expression levels after both treatments decreased to a lower level at 24 h. These results revealed that expression of the maize ZmMAG gene is induced by DNA-damaging agents, and suggest the plant MAG gene might function in DNA repair.

Real-time PCR analysis for the expression of ZmMAG in response to UV and zeocin treatments. Two-week-old maize seedlings grown in soil were collected for gene expression analysis of different time intervals. Total RNA was prepared from 2-week-old seedlings of wild-type maize after UV and zeocin treatments, respectively, and then reverse-transcribed. The resultant cDNAs were used as templates for real-time PCR analysis and ZmActin was used as an internal control. Real-time PCR was performed with ZmMAG specific primers and ZmActin specific primers. Data represent means and standard deviation of three replicates. Different letters following mean values indicate significant difference (P < 0.05, Tukey’s honestly significant difference test).
Discussion
More than a century ago, Charles Darwin derived and proposed the concept of the tree of life in the Origin of Species based on the hypothesis of gene mutation and vertical inheritance29. Since then, one of the goals of biologists is to develop the tree of life to describe the process of evolution30, 31. However, subsequent studies revealed that the genetic mechanisms do not only consist of genetic mutation and vertical inheritance, but also includes a novel mechanism, horizontal gene transfer (HGT). HGT plays important roles for the recipient organisms in acquiring new genetic material without the slow process of creating new genes by point mutation and recombination, and in acquiring new genetic material that may control important traits for adaptation to novel environments32. HGT, also known as lateral gene transfer (LGT), refers to the movement of genetic material between organisms with reproductive isolation other than via vertical inheritance (the transmission of DNA from parent to offspring)33,34,35. It was supposed that HGT is one of the most important forces driving the evolution of prokaryotes and certain unicellular eukaryotes, and leads to the spread of certain adaptive traits, such as antibiotic resistance and virulence32, 36, 37. Recent genomic surveys also found horizontally acquired genes in all major lineages of multicellular eukaryotes, including plants, fungi and animals, suggesting that HGT is also crucial for adaptive evolution throughout eukaryotic evolution38,39,40,41. The ubiquity of horizontally acquired genes in various cellular organisms leads to the concept of “web of life” rather than a progressively branching tree33, 42. Thus, detecting the genes acquired through horizontal transfer can not only provide understanding of molecular basis of adaptive evolution for the acceptor organisms, but can also provide references in building the “web of life”.
The biggest environmental change in the history of plant evolution is from aquatic to terrestrial environments. During their transition from water to land, plants had evolved some new phenotypic novelties and metabolic pathways to adapt to the terrestrial habit, including vascular tissues, stomata, cuticles, and the biosynthesis of plant polyamines and hormones43, 44. It was suggested that HGT played critically roles in the genomic innovations during the process of adaptive evolution for the colonization of land, such as the development of vascular tissues45, 46, the biosynthesis and degradation of some hormones47, 48 and disease resistance49. From water to terrestrial environments, one of the main environmental differences is that plants are directly exposed to UV and other DNA-damaging agents. Therefore, acquiring the genes that encode proteins for DNA repair is extremely important for the adaptive evolution of land plants.
The protein product of the bacterial MAG gene is thought to be involved in the recognition of a variety of base lesions and to initiate their repair via the BER pathway9, 16, 18, 19. In the present study, we found that the homologs of the MAG gene are widely present in streptophytes, from charophyte alga to angiosperm. At least two lines of evidence suggested that the Streptophyta MAG gene was acquired through an ancient horizontal gene transfer event from bacteria. First, all the green plant MAG genes formed a bootstrap-supported branch in the phylogeny, which is located in the clades of some bacteria such as gammaproteobacteria, deltaproteobacteria, and acidobacteria. Second, the protein products of the Streptophyta MAG genes showed the highest similarity to these bacterial homologs in this clade, and importantly, some motifs in the C-terminus are only conserved between land plants and these bacterial MAGs. We also noticed that the homologs of green plant MAG gene are present in Galdieria sulphuraria, a species of red alga. Because the green plants shared the same ancestor with red algae50, 51, the evolutionary scenario that the Streptophyta MAG gene was acquired from bacteria through HGT should exclude the possibility that this gene was inherited from the ancestor of green plants and red algae. In the phylogeny, the G. sulphuraria MAG gene was not located in the same branch with those in green plants, and more importantly, the protein product of this G. sulphuraria gene did not share any specifically evolutionarily conserved motifs with its homologs in streptophytes. Therefore, the most parsimonious explanation is that the origin of the Streptophyta MAG gene was the result of an ancient HGT event from bacteria, and the putative donators are gammaproteobacteria, deltaproteobacteria, and acidobacteria.
Charophyte algae, comprising in streptophytes together with land plants, are found in fresh water, with several ranging into brackish habitats, and several groups live in soils, crusts, and other aerial environments52. Comparison of the genome sequences with that of green algae and land plants demonstrate that charophyte algae acquired many genes specific to land plants, including those performing the functions in transcription regulation, signal transduction, stress responses, cell wall biogenesis and hormone-related functions53. In addition, it was also suggested that the most recent common ancestor of extant land plants and charophyte algae was preadapted for symbiotic associations54. In addition to land plants, the MAG gene is also present in the genomes of charophyte algae, such as K. flaccidum and P. margaritaceum. In the present study, we showed that the ancestor of charophyte algae and land plants had acquired the MAG gene through an ancient HGT event. In addition, two DNA-damaging agents, including UV radiation and zeocin, were selected to simulate DNA-damaging environments. UV radiation induces two of the most abundant mutagenic and cytotoxic DNA lesions such as cyclobutane–pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4 PPs) and their Dewar valence isomers9, while zeocin causes cell death by intercalating into DNA and induces double strand breaks of the DNA for most bacteria, fungi, plant, and animal cells55. Our experiments revealed that the expression of the maize ZmMAG gene was significantly induced by both UV radiation and zeocin, suggesting that plant MAG gene might be involved in the recognition and repair of a variety of base lesions. These results suggested that the ancestor of land plants acquired this gene through an HGT event for the preadaptation to DNA-damaging agents in terrestrial environments.
In general, prokaryotic genes contain no spliceosomal introns. However, nearly all green plant MAG genes contain five introns, and their positions and phases are highly conserved across all the lineages of green plants. This result suggested that these introns in Streptophyta MAG genes arose through insertions shortly after the HGT event and before the origin of the land plants. The phenomena that horizontally acquired genes further obtained introns has been observed in other HGT genes in plants44, 45. Because introns in eukaryotes fulfill a broad spectrum of functions, including virtually every step of mRNA processing, influencing and enhancing gene expression, and maintaining genomic structure56, 57, the quick acquisition of introns by the plant MAG gene shortly after the HGT event may represent the adaptive evolution to the acceptor genome and new environments. In our analysis, we found that the dominant driving force for MAG gene evolution in both streptophytes and bacteria (those in the same clade with plants) was purifying selection, which might contribute to functional stabilization58. However, when we used the bacterial genes as a background, positive selection was found to significantly contribute to the evolution of the Streptophyta MAG genes. Because point mutations under positive selection is a major force underlying the adaptation of organisms to a new environment59, the most reasonable explanation for these results is that the Streptophyta MAG gene may have acquired some functional innovations through positive selection shortly after the HGT event but before the separation of major land plant lineages. Although the actual function of the MAG gene in higher plants needs further investigation, some studies have also given some clues that the plant MAG gene may have gained putative functions other than its homologs in bacteria. For example, the expression of B. distachyon MAG gene was found to be regulated by salt stress26. In addition, the Arabidopsis AtMAG gene was preferentially expressed in meristematic tissue, the developing embryo and endosperm, and organ primordia, and strongly expressed in the mesophyll tissue of the growing leaf, suggesting that it might participate in the progress of cell growth60.
Conclusions
Because there are more DNA-damaging agents in terrestrial environments than in water, the origin and radiation of land plants should acquire new genes with the function of DNA repair. Therefore, understanding the evolutionary pattern of the genes involved in DNA repair will lay the foundation for revealing the genetic mechanism of the origin of land plants. In addition, it can also provide useful information for further genetic improvement of crop plants, because the stratospheric ozone layer, which can protect plant DNA from UV damaging, is continuously experiencing depletion. MAG has been shown to perform the functions in recognizing various base lesions in bacteria, and in initiating the process of the base excision repair for damaged DNA. In the present study, we provide clear evidence that the ancestor of land plants acquired the MAG gene through ancient HGT from bacteria. Further investigation revealed that the expression of the plant MAG gene is induced by DNA-damaging agents. The Streptophyta MAG genes had undergone positive selection during the initial evolutionary period after HGT, which might have led to the MAG obtaining functional innovation and fixation in the genome of the land plant ancestor. These findings revealed that the acquisition of MAG by the ancestor of land plants through HGT represents a preadaptation to terrestrial environments.
Materials and Methods
Sequence data sources
To identify the genes encoding MAG proteins in green plants, the protein sequence of the Arabidopsis gene AtMAG (AT3G12040)20, 60 was used as a query to search the NCBI nr and ref_seq protein, the spruce genome project61 and Phytozome62 databases. Then, the Pfam tool63 was used to predict the methylpurine-DNA glycosylase (Pur_DNA_glyco, PF02245) domain. The newly identified MAG sequences detected in green plants were used to search the respective sequence database iteratively. The deduced nucleotide and protein sequences of green plant MAG genes identified in this analysis were acquired from the Phytozome and NCBI nr and ref_seq databases. The redundant sequences were removed manually according to their chromosomal locations. To detect the MAG gene in Picea glauca, we searched the NCBI EST database using TBLASTN. Then, UniGene and ORFfinder were used to unite several ESTs into one full-length cDNA.
To identify the homologs of green plant MAG genes, BLAST searches against the nr protein sequence, NCBI EST and available eukaryotic genome databases were performed using the plant MAG protein sequences as queries. The obtained hits were further analyzed through a Pfam search to confirm the presence of the Pur_DNA_glyco domain. Protein sequences were sampled for a further combined phylogenetic analysis from representative groups within each domain of life (bacteria, archaea and eukaryotes) based on the BLASTP results.
Phylogenetic tree reconstruction
Multiple alignments for all the selected representative protein sequences were performed with Clustal X64. The gaps and ambiguously aligned sites were removed manually. Phylogenetic analyses were performed using maximum likelihood (ML) and neighbor-joining (NJ) methods using PhyML v3.065 and MEGA v7.066, respectively. The program ModelGenerator67 was used to identify the optimal model of protein substitution and rate heterogeneity. The ML phylogenetic analyses were conducted with the following parameters: Dayhoff model, estimated proportion of invariable sites, 4 rate categories, estimated gamma distribution parameter, and optimized starting BIONJ tree. The Jones-Taylor-Thornton (JTT) model was employed for the construction of NJ trees. A total of 100 non-parametric bootstrap samplings were carried out to estimate the support level for each internal branch for both the ML and NJ trees. The branch lengths and topologies of all phylogenies were calculated with PhyML.
Detection of selective constrains
To test the selective constrains of the MAG genes during the long period of evolution in both green plants and some selected bacteria, the values of the \({d}_{N}/{d}_{S}\) ratio (ω) for two groups of MAG genes were calculated with the program codeml in PAML v4.968. In this analysis, only bacterial MAG genes located in the same branch as green plant homologs were used. The PAL2NAL program69 was utilized for conversion of multiple alignment of proteins into the corresponding codon-based nucleotide alignment, which, in turn, was input into the codeml program in PAML. Three likelihood ratio tests (LRTs), including M0 vs. M3, M1a vs. M2a, and M7 vs. M8, were employed to examine the selective pressure. The LRT of the M0 vs. M3 comparison was used to test the heterogeneity in ω among the codon sites, while the other two LRTs were used to detect the role of positive selection. For one LRT, twice the difference of the log likelihood of the two models was compared with chi-square (χ 2) statistics, with degrees of freedom (DFs) equal to the difference in the number of parameters. In our analyses, the DFs were 3 for the M0/M3 LRT and 2 for the M1a/M2a and M7/M8 LRTs, respectively70, 71.
The improved branch-site model27 was also used to detect the effect of positive selection acting on the Streptophyta MAG genes following HGT. In these tests, we compared the null model (ω fixed to 1) with the alternative model (free ω). The branch of streptophytes was used as the foreground, while that containing the genes from bacteria, the putative donor of the Streptophyta MAG gene, was used as the background. Here, only the bacterial genes falling within the same branch as the Streptophyta genes were used. The Bayes empirical Bayes procedure28 in codeml was used to calculate the posterior probability that each site was subject to positive selection in the foreground branch.
Plant material, growth and stress treatments
Seeds of maize (Zea mays L.) inbred line B73 were surface-sterilized for 15 min in 0.05% sodium hypochlorite, and then washed thoroughly with sterile water. The sterilized seeds were germinated on filter paper saturated with distilled water in darkness for 2 days at 28 °C. For UV treatment, the uniformly germinated seeds were grown with sieved topsoil in the growth chamber, with a photoperiod of 14/10 h at 28/25 °C (light/night), 1200 μmol photons m−2 s−1 photosynthetically active radiation, and 70% relative humidity. Plants received 100 ml of water every day for 2 weeks. Two-week-old seedlings were moved to the UV light with 10 kJ m−2 d−1. The leaves of the seedlings were sampled at 0, 3, 6, 12, 24, 48 h after UV treatment, frozen in liquid nitrogen and stored at −80 °C for further analysis. For zeocin treatment, the uniformly germinated seeds were grown with culture solution in the growth chamber. The growth condition is the same as described above. Two-week-old seedlings were transferred to culture solutions containing 100 μM zeocin. The whole seedlings were sampled at 0, 3, 6, 12, 24, 48 h after zeocin treatment, frozen in liquid nitrogen and stored at −80 °C for further analysis.
RNA isolation and real-time PCR
Total RNA was extracted from frozen tissue using RNAsimple Total RNA Kit (TIANGEN) according to the manufacturer’s instructions. To check the ZmMAG expression level in maize plants in response to UV and zeocin treatment, approximately 2 μg of total RNA from each sample was used for first-strand cDNA synthesis by 5X All-In-One RT MasterMix (abm). Quantitative real-time PCR was performed with the EvaGreen 2X qPCR MasterMix (abm) with the following reaction conditions: 95 °C for 10 mins, then 30 cycles of 95 °C for 15 secs, 60 °C for 60 secs. The constitutively expressed ZmActin gene (GenBank accession no. J01238) was used as an internal control to normalize data. Quantitative gene expression was analyzed by comparative CT (Delta–Delta CT) method72. The primers were 5′-TTACGGGACCAGGAAAGGTTGG-3′ and 5′-AGGATGGTTCGACCAGTCAGTG-3′ for the ZmMAG gene, and 5′-GATGATGCGCCAAGAGCTG-3′ and 5′-GCCTCATCACCTACGTAGGCAT-3′ for ZmActin gene. All experiments were carried out using three biological replicates. The ZmMAG expression data were expressed as means ± standard deviation (SD). Statistical analysis was performed by one-way ANOVA and Tukey’s honestly significant difference test for significance (P < 0.05) by using IBM SPSS software (Version 22).
References
Finet, C., Timme, R. E., Delwiche, C. F. & Marleta, F. Multigene Phylogeny of the Green Lineage Reveals the Origin and Diversification of Land Plants. Current Biology 20, 2217–2222 (2010).
Zhong, B. J., Liu, L., Yan, Z. & Penny, D. Origin of land plants using the multispecies coalescent model. Trends in plant science 18, 492–495 (2013).
Qiu, Y. L. et al. The deepest divergences in land plants inferred from phylogenomic evidence. Proceedings of the National Academy of Sciences of the United States of America 103, 15511–15516 (2006).
Waters, E. R. Molecular adaptation and the origin of land plants. Molecular phylogenetics and evolution 29, 456–463 (2003).
Pires, N. D. & Dolan, L. Morphological evolution in land plants: new designs with old genes. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 367, 508–518 (2012).
Gill, S. S., Anjum, N. A., Gill, R., Jha, M. & Tuteja, N. DNA damage and repair in plants under ultraviolet and ionizing radiations. The Scientific World Journal 2015, 250158 (2015).
Tuteja, N., Singh, M. B., Misra, M. K., Bhalla, P. L. & Tuteja, R. Molecular mechanisms of DNA damage and repair: progress in plants. Critical reviews in biochemistry and molecular biology 36, 337–397 (2001).
Hu, Z., Cools, T. & De Veylder, L. Mechanisms Used by Plants to Cope with DNA Damage. Annual review of plant biology 67, 439–462 (2016).
Sinha, R. P. & Hader, D. P. UV-induced DNA damage and repair: a review. Photochemical & photobiological sciences: Official journal of the European Photochemistry Association and the European Society for Photobiology 1, 225–236 (2002).
Muller-Xing, R., Xing, Q. & Goodrich, J. Footprints of the sun: memory of UV and light stress in plants. Frontiers in plant science 5, 474 (2014).
Bray, C. M. & West, C. E. DNA repair mechanisms in plants: crucial sensors and effectors for the maintenance of genome integrity. The New phytologist 168, 511–528 (2005).
McCullough, A. K., Dodson, M. L. & Lloyd, R. S. Initiation of base excision repair: glycosylase mechanisms and structures. Annual review of biochemistry 68, 255–285 (1999).
Krokan, H. E., Standal, R. & Slupphaug, G. DNA glycosylases in the base excision repair of DNA. The Biochemical journal 325, 1–16 (1997).
David, S. S., O’Shea, V. L. & Kundu, S. Base-excision repair of oxidative DNA damage. Nature 447, 941–950 (2007).
Riazuddin, S. & Lindahl, T. Properties of 3-methyladenine-DNA glycosylase from Escherichia coli. Biochemistry 17, 2110–2118 (1978).
Wyatt, M. D., Allan, J. M., Lau, A. Y., Ellenberger, T. E. & Samson, L. D. 3-methyladenine DNA glycosylases: structure, function, and biological importance. BioEssays: news and reviews in molecular, cellular and developmental biology 21, 668–676 (1999).
Berdal, K. G., Johansen, R. F. & Seeberg, E. Release of normal bases from intact DNA by a native DNA repair enzyme. The EMBO journal 17, 363–367 (1998).
Trewick, S. C., Henshaw, T. F., Hausinger, R. P., Lindahl, T. & Sedgwick, B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178 (2002).
Connor, E. E. & Wyatt, M. D. Active-site clashes prevent the human 3-methyladenine DNA glycosylase from improperly removing bases. Chemistry & biology 9, 1033–1041 (2002).
Santerre, A. & Britt, A. B. Cloning of a 3-methyladenine-DNA glycosylase from Arabidopsis thaliana. Proceedings of the National Academy of Sciences of the United States of America 91, 2240–2244 (1994).
Malhotra, S. & Sowdhamini, R. Genome-wide survey of DNA-binding proteins in Arabidopsis thaliana: analysis of distribution and functions. Nucleic acids research 41, 7212–7219 (2013).
Fu, J. et al. Dissecting grain yield pathways and their interactions with grain dry matter content by a two-step correlation approach with maize seedling transcriptome. BMC plant biology 10, 63 (2010).
Wang, P. et al. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in Maize (Zea may). BMC genomics 16, 21 (2015).
Mak, Y. et al. A proteomic approach to the identification and characterisation of protein composition in wheat germ. Functional & integrative genomics 6, 322–337 (2006).
Tillett, R. L. et al. The Vitis vinifera C-repeat binding protein 4 (VvCBF4) transcriptional factor enhances freezing tolerance in wine grape. Plant biotechnology journal 10, 105–124 (2012).
Kim, D. Y., Hong, M. J., Jang, J. H. & Seo, Y. W. cDNA-AFLP analysis reveals differential gene expression in response to salt stress in Brachypodium distachyon. Genes Genom 34, 475–484 (2012).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular biology and evolution 22, 2472–2479 (2005).
Yang, Z., Wong, W. S. & Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Molecular biology and evolution 22, 1107–1118 (2005).
Darwin, C. The origin of species. (1872).
Ciccarelli, F. D. et al. Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287 (2006).
Hug, L. A. et al. A new view of the tree of life. Nature microbiology 1, 16048 (2016).
Ochman, H., Lawrence, J. G. & Groisman, E. A. Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304 (2000).
Soucy, S. M., Huang, J. & Gogarten, J. P. Horizontal gene transfer: building the web of life. Nat Rev Genet 16, 472–482 (2015).
Gogarten, J. P. & Townsend, J. P. Horizontal gene transfer, genome innovation and evolution. Nature reviews. Microbiology 3, 679–687 (2005).
Syvanen, M. Evolutionary implications of horizontal gene transfer. Annual review of genetics 46, 341–358 (2012).
de la Cruz, F. & Davies, J. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends in microbiology 8, 128–133 (2000).
Gluck-Thaler, E. & Slot, J. C. Dimensions of Horizontal Gene Transfer in Eukaryotic Microbial Pathogens. PLoS pathogens 11, e1005156 (2015).
Hotopp, J. C. D. Horizontal gene transfer between bacteria and animals. Trends Genet 27, 157–163 (2011).
Boto, L. Horizontal gene transfer in the acquisition of novel traits by metazoans. Proceedings. Biological sciences 281, 20132450 (2014).
Fitzpatrick, D. A. Horizontal gene transfer in fungi. FEMS microbiology letters 329, 1–8 (2012).
Yue, J., Hu, X. & Huang, J. Horizontal gene transfer in the innovation and adaptation of land plants. Plant signaling & behavior 8, e24130 (2013).
Qiu, H., Cai, G., Luo, J., Bhattacharya, D. & Zhang, N. Extensive horizontal gene transfers between plant pathogenic fungi. BMC biology 14, 41 (2016).
Ligrone, R., Duckett, J. G. & Renzaglia, K. S. Major transitions in the evolution of early land plants: a bryological perspective. Annals of botany 109, 851–871 (2012).
Yang, Z. et al. Evolution of land plant genes encoding L-Ala-D/L-Glu epimerases (AEEs) via horizontal gene transfer and positive selection. Bmc Plant Biol 13, 34 (2013).
Yang, Z. et al. Ancient horizontal transfer of transaldolase-like protein gene and its role in plant vascular development. The New phytologist 206, 807–816 (2015).
Tarrio, R., Ayala, F. J. & Rodriguez-Trelles, F. The Vein Patterning 1 (VEP1) gene family laterally spread through an ecological network. Plos One 6, e22279 (2011).
Yue, J., Hu, X. & Huang, J. Origin of plant auxin biosynthesis. Trends in plant science 19, 764–770 (2014).
Yue, J., Hu, X., Sun, H., Yang, Y. & Huang, J. Widespread impact of horizontal gene transfer on plant colonization of land. Nature communications 3, 1152 (2012).
Yang, Z. et al. Origin of the plant Tm-1-like gene via two independent horizontal transfer events and one gene fusion event. Scientific reports 6, 33691 (2016).
Rodriguez-Ezpeleta, N. et al. Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Current biology 15, 1325–1330 (2005).
Moreira, D., Le Guyader, H. & Philippe, H. The origin of red algae and the evolution of chloroplasts. Nature 405, 69–72 (2000).
Lewis, L. A. & McCourt, R. M. Green algae and the origin of land plants. American journal of botany 91, 1535–1556 (2004).
Hori, K. et al. Klebsormidium flaccidum genome reveals primary factors for plant terrestrial adaptation. Nature communications 5, 3978 (2014).
Delaux, P. M. et al. Algal ancestor of land plants was preadapted for symbiosis. Proceedings of the National Academy of Sciences of the United States of America 112, 13390–13395 (2015).
Chankova, S. G., Dimova, E., Dimitrova, M. & Bryant, P. E. Induction of DNA double-strand breaks by zeocin in Chlamydomonas reinhardtii and the role of increased DNA double-strand breaks rejoining in the formation of an adaptive response. Radiation and environmental biophysics 46, 409–416 (2007).
Le Hir, H., Nott, A. & Moore, M. J. How introns influence and enhance eukaryotic gene expression. Trends in biochemical sciences 28, 215–220 (2003).
Chorev, M. & Carmel, L. The function of introns. Frontiers in genetics 3, 55 (2012).
Yang, Z. et al. Adaptive evolution and divergent expression of heat stress transcription factors in grasses. BMC evolutionary biology 14, 147 (2014).
Przeworski, M., Coop, G. & Wall, J. D. The signature of positive selection on standing genetic variation. Evolution; international journal of organic evolution 59, 2312–2323 (2005).
Shi, L., Kent, R., Bence, N. & Britt, A. B. Developmental expression of a DNA repair gene in Arabidopsis. Mutation research 384, 145–156 (1997).
Nystedt, B. et al. The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584 (2013).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic acids research 40, D1178–1186 (2012).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic acids research 44, D279–285 (2016).
Chenna, R. et al. Multiple sequence alignment with the Clustal series of programs. Nucleic acids research 31, 3497–3500 (2003).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic biology 59, 307–321 (2010).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular biology and evolution 33, 1870–1874 (2016).
Keane, T. M., Creevey, C. J., Pentony, M. M., Naughton, T. J. & McLnerney, J. O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC evolutionary biology 6, 29 (2006).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution 24, 1586–1591 (2007).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic acids research 34, W609–612 (2006).
Nielsen, R. & Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–936 (1998).
Wong, W. S., Yang, Z., Goldman, N. & Nielsen, R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168, 1041–1051 (2004).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25, 402–408 (2001).
Acknowledgements
This work was supported by grants from the National Key Technology Research and Development Program of MOST (2016YFD0100300), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the National Natural Science Foundations (31391632, 91535103 and 31601810), the National High-tech R&D Program (863 Program) (2014AA10A601-5), the Natural Science Foundations of Jiangsu Province (BK20150010), the Natural Science Foundation of the Jiangsu Higher Education Institutions (14KJA210005), and the Innovative Research Team of Ministry of Agriculture.
Author information
Authors and Affiliations
Contributions
Z.Y., Y.Z., and C.X. conceived the study. H.F., L.H., R.C., P.L., S.X. and E.Z. performed the experiments. H.F., W.C., and L.L. analyzed the results. H.F., Y.Y. and G.L. prepared the figures. H.F., Z.Y., Y.Y., and L.H. wrote the paper. All authors reviewed the results and approved a final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, H., Huangfu, L., Chen, R. et al. Ancestor of land plants acquired the DNA-3-methyladenine glycosylase (MAG) gene from bacteria through horizontal gene transfer. Sci Rep 7, 9324 (2017). https://doi.org/10.1038/s41598-017-05066-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05066-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
