
Abstract
As a continuation of our efforts to discover and develop natural-product-based insecticidal agents, three novel and unusual 7-membered lactam derivatives of podophyllotoxin were prepared by thionyl chloride-mediated ring-expanded Beckmann rearrangement. The steric configurations of 3a–c were unambiguously identified by X-ray crystallography. It demonstrated that the configuration of the picropodophyllotoxin C4-oximes could also be confirmed by the corresponding C-ring expansion products via Beckmann rearrangement. Moreover, it was obviously further testified that when picropodophyllones reacted with hydroxylamine hydrochloride, only E configuration of picropodophyllotoxin C4-oximes was selectively produced. Compounds 3b and 3c showed more potent pesticidal activity than toosendanin against oriental armyworm, Mythimna separata (Walker).
Similar content being viewed by others
Introduction
Oriental armyworm, Mythimna separata Walker, is a typically polyphagous and gluttonous lepidopteran pest, and hard to control. Nowadays, the use of chemical insecticides continues to play an important role in the control of insect pests. However, the repeat and increasing application of those agrochemicals has led to the global dissemination of resistance in insect pests populations, and the serious human health and environmental problems1,2,3,4,5. In addition, the discovery and development of new insecticidal agents from plant secondary metabolites, or by using them as the lead compounds for further structural modifications, has recently been received much attention owing to their less or slower resistance development and low toxicity6,7,8.
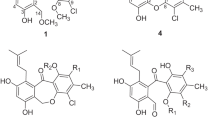
Podophyllotoxin (1, Fig. 1), isolated from the roots and rhizomes of Podophyllum hexandrum, is a naturally occurring aryltetralin lignan and contains five rings (labeled A–E). In addition to its mesmerizing structure, compound 1 has been used as the lead compound for preparation of potent anticancer drugs (e.g., etoposide, teniposide and etopophos)9,10,11, and insecticidal/antifungal agents12,13,14. More recently, we have prepared a series of 4α-acyloxy-2′(2′,6′)-(di)halogenopodophyllotoxins14 (I, Fig. 1), 2α-chloro-4α-acyloxy-2′(2′,6′)-(di)halogenopicropodophyllotoxins7 (II, Fig. 1), and oxime sulfonates of picropodophyllotoxin15 (III, Fig. 1)/ 2′(2′,6′)-(di)chloropicropodophyllotoxins16 (IV, Fig. 1) as insecticidal agents, and found some derivatives exhibited more potent insecticidal activity than toosendanin, a commercial botanical insecticide isolated from Melia azedarach. Especially it demonstrated that introduction of a chlorine atom at the C-2′ or C-2′,6′ position on the E-ring of picropodophyllotoxin was important for the insecticidal activity16. To the best of our knowledge, structural modification on the C-ring expansion of podophyllotoxins has not been carried out. In continuation of our program aimed at the discovery and development of new podophyllotoxin-based insecticidal agents, here we wanted to prepare unusual 7-membered lactam derivatives of podophyllotoxins by C-ring expansion reaction. Additionally, as shown in Fig. 1, the hydroxylamination products, picropodophyllotoxin C4-oximes (2, Eq. 1) and (2′, Eq. 2), might have E- and Z- configurations, respectively. Beckmann rearrangement is a well-known organic name reaction and its reaction mechanism has been fully proven17,18,19. According to the Beckmann rearrangement rule (Eq. 3, Fig. 1)17,18,19, the substituent R1 at the anti position to the hydroxyl group on the C=N moiety migrates to its nitrogen atom. Therefore, we envisioned that the configuration of the picropodophyllotoxin C4-oximes could also be confirmed by the corresponding C-ring expansion products via the Beckmann rearrangement.

Chemical structures of podophyllotoxin (1) and its derivatives (2, 2′, 3, 3′, and I-IV).
Materials and Instruments
All chemical reagents were purchased and utilized without further purification. Solvents were used directly or treated with standard methods before use. 2′-Chloropodophyllotoxin (4a) and 2′-bromopodophyllotoxin (4b) were all prepared in 85% yields (Fig. 2) according to our previous method14. Melting points (mp) were determined on a XT-4 digital melting point apparatus (Beijing Tech Instrument Co., Ltd., Beijing, China) and were uncorrected. Optical rotation was measured on a Rudolph Research Analytical Autopol III automatic polarimeter. Proton nuclear magnetic resonance spectra (1H NMR) was recorded in CDCl3 on a Bruker Avance 500 MHz instrument, and tetramethylsilane (TMS) was used as the internal standard.

Synthesis of lactam derivatives 3a–c by Beckmann rearrangement.
General procedure for synthesis of compounds 5a–5c
A mixture of compound 1, 4a or 4b (1.0 mmol), absolute ethanol (10 mL) and 10% aq. CH3CO2Na (10 mL) was refluxed, and the reaction process was checked by TLC analysis. After 15 h, the mixture was cooled at 0 °C and filtered to give the solid, which was further recrystallized from absolute ethanol to give 5a–5c.
Data for 5a: Yield = 75%, white solid, m.p. 222–223 °C; [α]20 D = +5 (c 3.2 mg/mL, CHCl3); 1H NMR (500 MHz, DMSO-d 6) δ: 7.06 (s, 1 H, H-5), 6.60 (s, 2 H, H-2′, 6′), 6.00 (s, 1 H, H-8), 5.91–5.95 (m, 3 H, H-1, OCH2O), 4.51 (d, J = 9.0 Hz, 1 H, H-4), 4.34–4.41 (m, 2 H, H-11), 3.92 (d, J = 7.5 Hz, 1 H, H-2), 3.74 (s, 6 H, 3′-OCH3, 5′-OCH3), 3.69 (s, 3 H, 4′-OCH3), 3.40–3.44 (m, 1 H, H-3).
Data for 5b: Yield = 90%, white solid, m.p. 116–117 °C; [α]20 D = +5 (c 3.2 mg/mL, acetone); 1H NMR (500 MHz, CDCl3) δ: 7.09 (s, 1 H, H-5), 6.61 (s, 1 H, H-8), 6.18 (s, 1 H, H-6′), 5.94 (s, 2 H, OCH2O), 4.66 (d, J = 9.5 Hz, 1 H, H-1), 4.47 (d, J = 9.5 Hz, 1 H, H-4), 4.40–4.43 (m, 1 H, H-11), 4.38 (d, J = 5.0 Hz, 1 H, H-11), 3.92 (s, 3 H, 3′-OCH3), 3.91 (s, 3 H, 5′-OCH3), 3.80 (s, 3 H, 4′-OCH3), 3.27–3.30 (m, 1 H, H-2), 2.90 (s, 1 H, 4-OH), 2.60–2.65 (m, 1 H, H-3).
Data for 5c: Yield = 83%, white solid, m.p. 82–84 °C; [α]20 D = −15 (c 3.4 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.08 (s, 1 H, H-5), 6.61 (s, 1 H, H-8), 6.21 (s, 1 H, H-6′), 5.93 (dd, J = 12.5, 1.0 Hz, 2 H, OCH2O), 4.65 (d, J = 9.5 Hz, 1 H, H-1), 4.51 (s, 1 H, H-4), 4.41-4.47 (m, 2 H, H-11), 3.92 (s, 3 H, 3′-OCH3), 3.91 (s, 3 H, 5′-OCH3), 3.80 (s, 3 H, 4′-OCH3), 3.30 (dd, J = 9.0, 6.0 Hz, 1 H, H-2), 2.63–2.67 (m, 1 H, H-3).
General procedure for synthesis of compounds 6a–6c
A mixture of 5a, 5b or 5c (0.5 mmol), chromium trioxide (CrO3, 2.5 mmol), and pyridine (5 mmol) in dry dichloromethane (15 mL) was stirred at room temperature. When the reaction was complete after 3 h, checked by TLC analysis, the mixture was diluted by dichloromethane, washed by saturated aq. NaHSO3 and brine, dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by silica gel column chromatography to afford 6a–6c.
Data for 6a: Yield = 94%, white solid, m.p. 152–154 °C; [α]20 D = −164 (c 3.4 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.49 (s, 1 H, H-5), 6.68 (s, 1 H, H-8), 6.23 (s, 2 H, H-2′, 6′), 6.04 (d, J = 3.5 Hz, 2 H, OCH2O), 4.75 (d, J = 9.0 Hz, 1 H, H-11), 4.69 (s, 1 H, H-1), 4.33-4.36 (m, 1 H, H-11), 3.80 (s, 3 H, 4′-OCH3), 3.75 (s, 6 H, 3′, 5′-OCH3), 3.30–3.32 (m, 2 H, H-2, 3).
Data for 6b: Yield = 86%, white solid, m.p. 105–107 °C; [α]20 D = −30 (c 3.4 mg/mL, acetone); 1H NMR (500 MHz, CDCl3) δ: 7.51 (s, 1 H, H-5), 6.68 (s, 1 H, H-8), 6.07 (d, J = 1.0 Hz, 2 H, OCH2O), 5.85 (s, 1 H, H-6′), 5.21 (d, J = 1.5 Hz, 1 H, H-1), 4.75 (d, J = 9.0 Hz, 1 H, H-11), 4.32–4.35 (m, 1 H, H-11), 3.95 (s, 3 H, 3′-OCH3), 3.85 (s, 3 H, 5′-OCH3), 3.54 (s, 3 H, 4′-OCH3), 3.42 (dd, J = 8.0, 2.0 Hz, 1 H, H-2), 3.15–3.18 (m, 1 H, H-3).
Data for 6c: Yield = 80%, white solid, m.p. 98–100 °C; [α]20 D = −53 (c 3.2 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.51 (s, 1 H, H-5), 6.67 (s, 1 H, H-8), 6.06 (d, J = 8.5 Hz, 2 H, OCH2O), 5.88 (s, 1 H, H-6′), 5.22 (s, 1 H, H-1), 4.74 (d, J = 9.5 Hz, 1 H, H-11), 4.32–4.35 (m, 1 H, H-11), 3.93 (s, 3 H, 3′-OCH3), 3.84 (s, 3 H, 5′-OCH3), 3.53 (s, 3 H, 4′-OCH3), 3.43 (dd, J = 8.0, 1.5 Hz, 1 H, H-2), 3.14–3.16 (m, 1 H, H-3).
General procedure for synthesis of compounds 2a–2c
A mixture of 6a, 6b or 6c (0.53 mmol), hydroxylamine hydrochloride (0.8 mmol), and pyridine (2.12 mmol) in absolute ethanol (20 mL) was refluxed, and the reaction process was checked by TLC analysis. After 72 h, the solvent was removed under reduced pressure and saturated aq. NaHCO3 was added to the residue, which was extracted with ethyl acetate. The combined organic phase was dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography to afford 2a–2c.
Data for 2a: Yield = 80%, White solid, m.p. 114–116 °C; [α]20 D = −53 (c 3.0 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.27 (s, 1 H, H-5), 6.69 (s, 1 H, H-8), 6.25 (s, 2 H, H-2′, 6′), 5.98 (d, J = 2.5 Hz, 2 H, OCH2O), 4.57 (d, J = 2.0 Hz, 1 H, H-1), 4.51–4.52 (m, 2 H, H-11), 3.98–4.02 (m, 1 H, H-3), 3.79 (s, 3 H, 4′-OCH3), 3.74 (s, 6 H, 3′, 5′-OCH3), 3.24 (dd, J = 2.5, 8.5 Hz, 1 H, H-2).
Data for 2b: Yield = 83%, white solid, m.p. 106–107 °C; [α]20 D = −27 (c 3.4 mg/mL, acetone); 1H NMR (500 MHz, CDCl3) δ: 7.27 (s, 1 H, H-5), 6.70 (s, 1 H, H-8), 5.99 (s, 2 H, OCH2O), 5.95 (s, 1 H, H-6′), 5.07 (d, J = 2.0 Hz, 1 H, H-1), 4.47–4.53 (m, 2 H, H-11), 3.93 (s, 3 H, 3′-OCH3), 3.83–3.86 (m, 4 H, H-3, 5′-OCH3), 3.52 (s, 3 H, 4′-OCH3), 3.43 (dd, J = 8.5, 2.5 Hz, 1 H, H-2).
Data for 2c: Yield = 82%, white solid, m.p. 124–126 °C; [α]20 D = −32 (c 3.9 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.28 (s, 1 H, H-5), 6.69 (s, 1 H, H-8), 6.00 (s, 1 H, H-6′), 5.99 (s, 2 H, OCH2O), 5.07 (d, J = 2.5 Hz, 1 H, H-1), 4.46–4.52 (m, 2 H, H-11), 3.91 (s, 3 H, 3′-OCH3), 3.83–3.86 (m, 4 H, H-3, 5′-OCH3), 3.54 (s, 3 H, 4′-OCH3), 3.43 (d, J = 8.5 Hz, 1 H, H-2).
General procedure for synthesis of compounds 3a–3c
A solution of thionyl chloride (0.63 mL) in tetrahydrofuran (5 mL) was added dropwise to a solution of 2a, 2b or 2c (0.25 mmol) in tetrahydrofuran (5 mL) at 0 °C. Then the above mixture was stirred, and the reaction temperature was gradually raised to room temperature. The reaction process was checked by TLC analysis. After 72 h, water was added to the mixture, and its pH value was adjusted with ammonia to 8-9. Subsequently, the mixture was extracted with dichloromethane. The combined organic phase was washed by water, 5% aq. NaHCO3 and brine, dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by preparative thin-layer chromatography (PTLC) to produce 3a–3c.
Data for 3a: Yield = 21%, white solid, m.p. 232–234 °C; [α]20 D = −85 (c 3.1 mg/mL, CHCl3); IR cm−1 (KBr): 3443, 2930, 2844, 1767, 1680, 1594, 1493, 1474, 1240, 1130, 1039; 1H NMR (500 MHz, CDCl3) δ: 8.29 (s, 1 H, NH), 6.56 (s, 1 H), 6.47 (brs, 2 H), 6.29 (s, 1 H), 5.94 (dd, J = 8.0, 1.0 Hz, 2 H, OCH2O), 4.99 (brs, 1 H, H-1), 4.37 (d, J = 12.0 Hz, 1 H, H-11), 4.21 (dd, J = 8.5, 6.0 Hz, 1 H, H-2), 3.87 (s, 4 H, OCH3, H-11), 3.83 (s, 6 H, 2 × OCH3), 3.38 (brs, 1 H, H-3); HRMS (ESI): Calcd for C22H22O8N ([M + H]+), 428.1340; Found, 428.1344.
Data for 3b: Yield = 18%, white solid, m.p. 155–157 °C; [α]20 D = −141 (c 2.1 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.57 (s, 1 H), 6.69 (s, 1 H), 6.57 (s, 1 H), 6.09 (brs, 1 H, H-1), 5.94 (d, J = 6.0 Hz, 2 H, OCH2O), 5.11 (d, J = 9.0 Hz, 1 H, H-11), 4.83 (d, J = 13.0 Hz, 1 H, H-11), 4.21 (dd, J = 9.5, 5.5 Hz, 1 H, H-2), 3.94 (s, 3 H, 3′-OCH3), 3.90 (s, 3 H, 4′-OCH3), 3.89 (s, 3 H, 5′-OCH3), 3.35 (dd, J = 9.5, 5.5 Hz, 1 H, H-3); HRMS (ESI): Calcd for C22H21O8NCl ([M + H]+), 462.0950; Found, 462.0949.
Data for 3c: Yield = 19%, white solid, m.p. 158–160 °C; [α]20 D = −116 (c 2.9 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.68 (s, 1 H), 6.72 (s, 1 H), 6.57 (s, 1 H), 6.06 (s, 1 H, H-1), 5.94 (dd, J = 5.5, 1.0 Hz, 2 H, OCH2O), 5.11 (d, J = 9.5 Hz, 1 H, H-11), 4.84 (d, J = 13.0 Hz, 1 H, H-11), 4.20 (dd, J = 9.0, 5.5 Hz, 1 H, H-2), 3.94 (s, 3 H, 3′-OCH3), 3.90 (s, 3 H, 4′-OCH3), 3.89 (s, 3 H, 5′-OCH3), 3.34 (dd, J = 9.5, 5.5 Hz, 1 H, H-3); 13C NMR (125 MHz, CDCl3) δ: 173.0, 169.8, 152.5, 151.6, 147.1, 146.1, 142.5, 132.1, 129.4, 128.9, 112.8, 108.2, 107.6, 104.1, 101.7, 65.4, 61.1, 61.0, 56.4, 49.9, 42.9, 29.7; HRMS (ESI): Calcd for C22H21O8NBr ([M + H]+), 506.0445; Found, 506.0441.
Biological assay
The insecticidal activity of 1–6 was tested as the mortality rate values by using the leaf-dipping method14, against the pre-third-instar larvae of oriental armyworm, Mythimna separata (Walker). Toosendanin, isolated from Melia azedarach, was used as a positive control and supplied by Research & Development Center of Biorational Pesticide, Northwest A&F University, Shaanxi province, China. For each compound, 30 larvae (10 larvae per group) were used. Acetone solutions of all tested compounds, and toosendanin were prepared at the concentration of 1 mg/mL. Fresh wheat leaves were dipped into the corresponding solution for 3 s, then taken out, and dried in a room. Leaves treated with acetone alone were used as a blank control group. Several treated leaves were kept in each dish, where every 10 larvae were raised. If the treated leaves were consumed, additional treated leaves were added to the dish. After 48 h, compound-soaked leaves were removed, and untreated fresh ones were added to all dishes till adult emergence. The experiment was carried out in a conditioned room (25 ± 2 °C, 65–80% relative humidity (RH), 12 h/12 h (light/dark) photoperiod). The insecticidal activity of the tested compounds against M. separata was calculated by the formula
Where T is the mortality rate in the group treated with the tested compounds, and C is the mortality rate in the blank control group (T and C were all expressed as the percentage).
Results and Discussion
As shown in Fig. 2, 2′-chloropodophyllotoxin (4a) and 2′-bromopodophyllotoxin (4b) were smoothly prepared by reaction of 1 with 1.1 equiv of N-chlorosuccinimide (NCS) and N-bromosuccinimide (NBS), respectively14. Then, picropodophyllotoxin (5a), 2′-chloropicropodophyllotoxin (5b) and 2′-bromopicropodophyllotoxin (5c) were obtained by reaction of 10% aq. NaOAc with 1, 4a and 4b, respectively15, 16. Subsequently, oxidation of 5a–c in the presence of chromium trioxide (CrO3) and pyridine afforded picropodophyllones (6a–c)15, 16. When 6a–c further reacted with hydroxylamine hydrochloride, only picropodophyllotoxin C4-oximes 2a–c (E configuration), testified in our previous paper15, 16, were selectively obtained, whereas their Z configuration isomers 2′a–c were not detected. Finally, in the presence of thionyl chloride, three novel 7-membered lactam derivatives of podophyllotoxin (3a–c) were produced via Beckmann rearrangement of the C-ring of 2a–c. The steric configurations of 3a–c were unambiguously identified by X-ray crystallography (Figs 3–5). It clearly demonstrated that the NH groups of 7-membered lactams of 3a–c were connected with their B-ring (phenyl ring). Based on the Beckmann rearrangement rule17,18,19, the substituent at the anti position to the hydroxyl group on the C=N moiety migrates to its nitrogen atom. Therefore, the phenyl ring should be at the anti position to the hydroxyl group, and it further demonstrated that picropodophyllotoxin C4-(E) oximes 2a–c was selectively produced. Three crystallographic data (excluding structure factors) for the structures of 3a–c, in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1495773, 1495719, and 1495720, respectively. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44 (0)1223 336033 or e-mail: deposit@ccdc.cam.ac.uk].

The X-ray crystal structure of 3a. Drawing by Hui Xu.

The X-ray crystal structure of 3b. Drawing by Hui Xu.

The X-ray crystal structure of 3c. Drawing by Hui Xu.
Moreover, according to the Beckmann rearrangement rule17,18,19, the mechanism for synthesis of 3a–c by thionyl chloride-mediated Beckmann rearrangement was described in Fig. 6. First, thionyl chloride activated the oxime fragments of 2a–c to give 7a–c. Then the phenyl (B-ring) at an anti position to the hydroxyl group migrated to the oxime nitrogen atom of 7a–c, and the corresponding key nitrilium cations 8a–c were produced. Finally, compounds 8a–c were further hydrolyzed to 3a–c via the intermediates 9a–c.

Mechanism for synthesis of 7-membered lactams 3a–c by thionyl chloride-mediated Beckmann rearrangement.
As shown in Table 1, the insecticidal activity of compounds 1-6 against the pre-third-instar larvae of M. separata in vivo was tested by the leaf-dipping method at a concentration of 1 mg/mL. Toosendanin, a commercial botanical insecticide isolated from Melia azedarach, was used as the positive control at 1 mg/mL. Leaves treated with acetone alone were used as a blank control group. The corresponding mortality rates of the treated groups after 35 days were higher than those after 10 and 20 days in the same as in our previous papers14,15,16. For example, the corrected mortality rates of 3b against M. separata after 10 and 20 days were 10% and 16.7%, respectively. However, it was remarkablely increased to 56.7% after 35 days, which was more than 5-fold of that after 10 days. Among all derivatives, two lactams 3b and 3c exhibited more potent insecticidal activity than the positive control toosendanin. For example, the final mortality rates (FMRs) of 3b and 3c were 56.7% and 60.0%, respectively; whereas the FMRs of the precursor 1 and toosendanin were 33.3% and 50.0%, respectively. It further demonstrated that introduction of a halogen atom at the C-2′ position on the E-ring of picropodophyllotoxin or podophyllotoxin was important for the insecticidal activity14, 16. For example, the FMRs of 2b and 2c were 33.3% and 43.3%, respectively; whereas the FMRs of 2a was 23.3%; the FMRs of 4a and 4b were 50.0% and 46.7%, respectively; whereas the FMRs of 1 was 33.3%; the FMRs of 5b and 5c were 50.0% and 43.3%, respectively; whereas the FMRs of 5a was 36.7%; the FMRs of 6b and 6c were 36.7% and 46.7%, respectively; whereas the FMRs of 6a was 30.0%. Finally, to obtain the EC50 value, further biological assay for compound 3c was conducted; the EC50 value of compound 3c was 0.809 mg/mL (See Supporting Information).
Conclusion
In summary, three novel and unusual 7-membered lactam derivatives of podophyllotoxin were obtained by thionyl chloride-mediated ring-expanded Beckmann rearrangement of picropodophyllotoxin C4-oximes. The steric configurations of 3a–c were all unambiguously confirmed by X-ray crystallography. It further demonstrated that when picropodophyllones reacted with hydroxylamine hydrochloride, only picropodophyllotoxin C4-(E) oximes were selectively produced. Especially compounds 3b and 3c showed more potent insecticidal activity than toosendanin against M. separata. It showed that introducing a halogen atom at the C-2′ position on the E-ring of picropodophyllotoxin/podophyllotoxin was important for the insecticidal activity. It will pave the way for further design and structural modifications of podophyllotoxin as botanical insecticidal agents.
References
Heckel, D. G. Insecticide resistance after silent spring. Science 337, 1612–1614 (2012).
Guillette, L. J. Jr. & Iguchi, T. Life in a contaminated world. Science 337, 1614–1615 (2012).
Seong, K. M., Sun, W., Clark, J. M. & Pittendrigh, B. R. Splice form variant and amino acid changes in MDR49 confers DDT resistance in transgenic Drosophila. Sci. Rep. 6, 23355 (2016).
Seiber, J. N., Coats, J., Duke, S. O. & Gross, A. D. Biopesticides: state of the art and future opportunities. J. Agric. Food Chem. 62, 11613–11619 (2014).
Edwards, K. T., Caprio, M. A., Allen, K. C. & Musser, F. R. Risk assessment for Helicoverpa zea (Lepidoptera: Noctuidae) resistance on dual-gene versus single-gene corn. J. Econom. Entomol. 106, 382–392 (2013).
Qu, H., Lv, M., Yu, X., Lian, X. & Xu, H. Discovery of some piperine-based phenylsulfonylhydrazone derivatives as potent botanically narcotic agents. Sci. Rep. 5, 13077 (2015).
Fan, L. L., Zhi, X. Y., Che, Z. P. & Xu, H. Insight into 2α-chloro-2′(2′,6′)-(di)halogenopicropodophyllotoxins reacting with carboxylic acids mediated by B.F3·Et2O. Sci. Rep 5, 16285 (2015).
Dayan, F. E., Cantrell, C. L. & Duke, S. O. Natural products in crop protection. Bioorg. Med. Chem. 17, 4022–4034 (2009).
You, Y. J. Podophyllotoxin derivatives: Current synthetic approaches for new anticancer agents. Curr. Pharm. Des 11, 1695–1717 (2005).
Gordaliza, M., Garcia, P. A., Miguel Del Corral, J. M., Castro, M. A. & Gomez-Zurita, M. A. Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives. Toxicon 44, 441–459 (2004).
Lv, M. & Xu, H. Recent advances in semisynthesis, biosynthesis, biological activities, mode of action, and structure-activity relationship of podophyllotoxins: an update (2008-2010). Mini-Rev. Med. Chem. 11, 901–909 (2011).
Kumar, K. A., Singh, S. K., Kumar, B. S. & Doble, M. Synthesis, anti-fungal activity evaluation and QSAR studies on podophyllotoxin derivatives. Cent. Eur. J. Chem. 5, 880–897 (2007).
Miyazawa, M., Fukuyama, M., Yoshio, K., Kato, T. & Ishikawa, Y. Biologically active components against Drosophila melanogaster from Podophyllum hexandrum. J. Agric. Food Chem. 47, 5108–5110 (1999).
Che, Z. P., Yu, X., Zhi, X. Y., Fan, L. L. & Xu, H. Synthesis of novel 4α-(acyloxy)-2′(2′,6′)-(di)halogenopodophyllotoxin derivatives as insecticidal agents. J. Agric. Food Chem. 61, 8148–8155 (2013).
Wang, Y. et al. Synthesis and quantitative structure-activity relationship (QSAR) study of novel isoxazoline and oxime derivatives of podophyllotoxin as insecticidal agents. J. Agric. Food Chem. 60, 8435–8443 (2012).
Wang, R., Zhi, X. Y., Li, J. & Xu, H. Synthesis of novel oxime sulfonate derivatives of 2′(2′,6′)-(di)chloropicropodophyllotoxins as insecticidal agents. J. Agric. Food Chem. 63, 6668–6674 (2015).
Lutz, J., M., R., Zeller, M. & Becker, D. P. Beckmann rearrangement of cyclotriveratrylene (CTV) oxime: tandem Beckmann-electrophilic aromatic addition. Tetrahedron. Lett. 49, 5003–5005 (2008).
Hashimoto, M., Obora, Y., Sakaguchi, S. & Ishii, Y. Beckmann rearrangement of ketoximes to lactams by triphosphazene catalyst. J. Org. Chem. 73, 2894–2897 (2008).
De Luca, L., Giacomelli, G. & Porcheddu, A. Beckmann rearrangement of oximes under very mild conditions. J. Org. Chem. 67, 6272–6274 (2002).
Acknowledgements
The present research was partly supported by National Natural Science Foundation of China (No. 31672071), and Special Funds of Central Colleges Basic Scientific Research Operating Expenses (No. 2452015096).
Author information
Authors and Affiliations
Contributions
X.Z. and Y.Z. performed experiments, and analysed data; J.H. performed experiments; H.X. designed experiments, analysed data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhi, X., Zhang, Y., Huang, J. et al. Seven-Membered Lactam Derivatives of Podophyllotoxins as New Pesticidal Agents. Sci Rep 7, 3917 (2017). https://doi.org/10.1038/s41598-017-04136-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04136-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
