
Abstract
Diarrhoea lasting longer than 14 days and failing to respond to conventional management is defined as severe and protracted diarrhoea (SD). In this study, we investigated the prevalence, pathogens and prognosis of SD in primary immunodeficiency diseases (PIDs). Among 246 patients with predominantly paediatric-onset PIDs from 2003–2015, 21 [Btk (six), IL2RG (four), WASP, CD40L, gp91 (three each), gp47, RAG2 (one each)] and five [CVID (four), SCID (one)] without identified mutations had SD before prophylactic treatment. Detectable pathogens included pseudomonas, salmonella (six each), E. coli, cytomegalovirus, coxsackie virus and cryptosporidium (one each), all of whom improved after a mean 17 days of antibiotics and/or IVIG treatment. Seven (7/26; 27.0%) patients died [respiratory failure (four), lymphoma, sepsis and intracranial haemorrhage (one each)]. The patients with WAS, CGD and CD40L and SD had a higher mortality rate than those without. Another five males with mutant XIAP, STAT1, FOXP3 (one each) and STAT3 (two) had undetectable-pathogenic refractory diarrhoea (RD) that persisted >21 days despite aggressive antibiotic/steroid treatment and directly resulted in mortality. For the patients with RD without anti-inflammatory optimization, those with mutant XIAP and FOXP3 died of Crohn’s-like colitis and electrolyte exhaustion in awaiting transplantation, while transplantation cured the STAT1 patient.
Similar content being viewed by others


Introduction
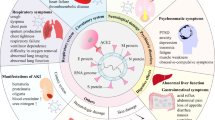
The intestinal tract is the largest lymphoid organ in the body and contains lymphocytes, macrophages, and dendritic cells to prevent pathogenic invasion, modulate inflammation1, 2, and produce sufficient antibodies for optimal neutralization, opsonization, and complement activation3, 4. If these cellular and humoral immune responses do not effectively cooperate and mutually balance, unwanted or excess reactions will injure the epithelium, mucosa, submucosa, and connective tissue, consequently leading to persistent diarrhoea despite nothing per os, hydration, antibiotics and immunosuppressant therapy1,2,3.
Severe and protracted diarrhoea (SD) is generally defined as prolonged diarrhoea lasting for more than 2 weeks, usually emerging within 2 years of life and requiring parenteral nutrition5, although the definition has undergone several revisions to better delineate the duration and clinical course6. Some studies have emphasised that specific aetiologies including infections that persist for longer than expected should be included in the definition.
Excluding congenital enterocyte defects7, 8, SD is often accompanied by “little effect of treatment”, “failure to gain weight”, and/or co-morbidity with “recurrent infections”, all of which are warning signs of primary immunodeficiency diseases (PIDs)9. Increasing evidence suggests that SD can mimic inflammatory bowel disease (IBD) and that it is associated with PIDs patients who have defective IL-12/2310,11,12,13,14,15,16 and IL-10 signalling17,18,19,20,21, profound T cell defects22,23,24,25,26,27,28,29, nicotinamide adenine dinucleotide phosphate oxidase anomalies30, 31, anti-apoptosis signalling of X-linked inhibitor of apoptosis (XIAP)32,33,34,35, and nuclear factor kappa B transcription (NEMO) signalling36. Research linking SD to monogenetic PIDs has shed light on the pathogenesis, and suggests that hematopoietic stem cell transplantation (HSCT) can be a potential cure, especially if there is profound T and polymorphonuclear cell deficiencies leading to recurrent opportunistic life-threatening infections and serious persistent intestinal inflammation.
With increased susceptibility to infections and inflammation, PIDs patients can develop the SD phenotype and present with frequent diarrhoea for longer than 14 days despite conventional management1, 3. In this nationwide study from the referral institute of Primary Immunodeficiency Care and Research (PICAR), we investigated the prevalence, pathogens and prognosis of PIDs patients with the SD phenotype.
Results
Enrolled Patients
Of the enrolled patients, 26 (2 females) had the SD phenotype, including the Btk gene in six, IL2RG in four, WASP in three, CD40L in three, gp91 in three, gp47 in one, RAG2 in one, and four with CVID and one with SCID without identified genetic defects. The mutations included splicing, deletion, missense and stop mutations, but not insertion mutations (Table 1). Their median age at onset of the SD phenotype was 14 months (range 2–273 months; mean ± S.D. 48.6 ± 65.0 months), with a median duration of diarrhoea of 14 days (range 14–18 days; mean 15.0 ± 1.6 days), median length of total parental nutrition (TPN) of 7 days (range 4–10 days; mean 7.0 ± 2.0 days), and median length of antibiotic treatment of 16.5 days (range 16–20 days; mean 16.9 ± 1.7 days) including ceftriaxone, a combination of ceftazidime and amikacin, and carbapenems and/or meropenem. Neutropenia was noted in four patients (Btk1-2, Btk3, Btk5 and SCID2-IL2RG) with pseudomonas colitis and two (SCID3-IL2RG and CGD4-gp47) with salmonella colitis. These patients also developed bronchiectasis (11), sepsis (7), and significant other events (21) before adequate prophylactic treatment which included sulfamethoxazole/trimethoprim and fluconazole for T cell deficiency, sulfamethoxazole/trimethoprim, itraconazole, and/or interferon-gamma for polymorphonuclear cell deficiency, regular immunoglobulin infusion for B cell deficiency, and ampicillin-sulbactam for bronchiectasis.
Another five male patients had a poor response to intravenous immunoglobulin (IVIG) and empiric antibiotics plus steroids over 3 weeks, and were defined as having the refractory diarrhoea (RD) phenotype (Table 2) with blood-stained diarrhoea but without obvious protein-losing enteropathy. Four patients had a peri-anal abscess without fistula formation, and one (patient 4) had a perforated colon and severe pyoderma in the abdominal wall that damaged his rectus muscularis. Peritoneal reconstruction with a 10 × 10 cm skin graft was performed to cover the injured abdominal wall, however he was complicated by an intestinal peritoneal cutaneous fistula (Fig. 1). Anaemia, elevated ESR, and hypoalbuminemia indicated chronic inflammation, consistent with the coloscopic observation (Fig. 2) and the pathological findings (Figs 3 and 4).

The morbidity of refractory severe and protracted diarrhoea (RD) in patient 4 was caused by a cutaneous peritoneal intestinal fistula.

Colonofiberoscopy revealed a cobblestone mucosal pattern and multiple pseudo-polyp-like lesions.

(A) Small intestine biopsy of patient 1 revealed marked villous atrophy and acute and chronic inflammation (hematoxylin-eosin [H&E] staining, magnification 100X) and (B) granulomatous inflammation (white arrows) composed of aggregates of epithelioid histiocytes in the lamina propria (H&E, 200X). (C,D) Colon biopsy of patient 1 showed ulceration and acute and chronic inflammation, with prominent epithelial apoptosis (white arrows) and intra-epithelial neutrophils (H&E, 400X).

An endoscopic biopsy in patient 2 demonstrated (A) moderate lymphocytic infiltrates in the oesophagus (H&E staining, 200X) and (B) paucity of plasma cells in the stomach (H&E, 400X). (C) In patient 3, repeated colonic biopsies disclosed mildly increased lymphocytes and decreased plasma cells without granuloma, ulcer, or cryptitis (H&E, 400X). (D) A gastric biopsy in patient 5 revealed chronic active gastritis and colonization of Helicobacter pylori (H&E, 200X).
Immunologic Studies and Genetic Analysis
The characteristics of PIDs patients with the SD phenotype (Table 1) including micro-thrombocytopenia, recurrent sinopulmonary infections, hypogammaglobulinemia, catalase-positive pathogens, opportunistic infections, and lymphopenia were recognized as Wiskott-Aldrich syndrome (WAS), X-linked agammaglobulinemia (XLA), X-linked hyper IgM syndrome (HIGM), common variable immunodeficiency (CVID), chronic granulomatous disease (CGD), severe combined T- and B-cell immunodeficiency (SCID), according to the genetic mutations of the updated PIDs classification37, 38.
The immunologic evaluation of these five patients with RD (Table 2) showed that one had neutropenia, one had decreased absolute lymphocytes, naïve CD4, and CD8 cells, two had decreased memory CD4 cells, and four had decreased memory CD19 B cells. Quantitative levels of IgE were increased in four patients and the level of IgA was increased in one. Lymphocyte proliferation was decreased in two patients, while four had impaired liver function. Superoxide production from stimulated PMNs and suppression of TNF-α production through IL-10 signalling in LPS-treated PBMCs were within normal range.
Of note, patient 1 also had extreme splenomegaly and recurrent hemophagocytic lymphohistiocytosis with a high EBV viral load32,33,34,35. He had a splicing mutation at intron 5 (+1) G > A (Supplementary Fig. 1A), missing exon 5, frameshift at the 354th Val ending at the 379th due to the lack of the RING (Really Interesting New Gene) domain in the XIAP gene (Supplementary Fig. 1B) to inhibit caspase 3 or 7 activity. His mother was a carrier. Patient 2 had a STAT1 mutation impairing the IFN-γ-IL-12/23 circuit, thereby increasing susceptibility to mycobacterial and Candida infections. A [Thr385Met] STAT1 mutation in the DNA-binding domain region also caused an IPEX-like phenotype (immune dysregulation, polyendocrinopathy, enteropathy, and X-linked)39,40,41. This genetic mutation was finally identified 8 years post-transplantation (Supplementary Fig. 1C). Patient 3 had the IPEX phenotype, which had caused the death of his maternal uncle due to diabetes. His [Met370Leu] FOXP3 mutation changed the fork-head domain and decreased the percentage of Treg cells (CD25 + + CD4+/CD4+, 5.8% vs. 32.6%) (Supplementary Fig. 1D and E). In patients 4 and 5, extremely high IgE levels and recurrent cutaneous infections were the distinct manifestations of hyper IgE syndrome. The STAT3 mutations were found to have IVS 10 (−2) A > G leading to a splicing loss of exon 11 and missense mutation of Gln469Arg in exon 15 (Supplementary Fig. 1F and G, respectively, in the DNA-binding domain region). Both were de novo loss-of-function mutations and their family members did not have the hyper IgE syndrome phenotype.
Treatment Response and Prognosis
Salmonella and pseudomonas strains were the most common of 17 identified pathogens in the patients with the SD phenotype (in 6 and 6 patients, respectively). Even though cytomegalovirus and cryptosporidium were only isolated in one patient with SCID4-IL2RG and one with HIGM1-CD40L, both had persistent cramping pain and high CRP levels, implying possibly mixed bacterial pathogens in such opportunistic infections. Both patients improved with effective antibiotic treatment plus gancyclovir and nitazoxanide, respectively. Giardia and clostridium difficile toxin A were not detected in any of our patients. In addition to IVIG infusion for those with hypogammaglobulinemia according to age, those with the SD phenotype improved within 3 weeks by effective and empritic antibiotics. However, even though all of the patients with RD without hypogammaglobulinemia received consistent treatment including IVIG and empiric antibiotics plus methylprednisolone over 3 weeks, only a limited effect was achieved. The exception was patient 4 who had a STAT3 mutation, in whom the number of RD flare-ups decreased from 5–6 episodes per year to 1–2 episodes per year after receiving steroids and regular IVIG treatment. During his episodes, the frequency was an average 5–6 times per day lasting for 15–22 days.
Seven of the 26 patients (27.0%) with SD died of respiratory failure (four), and lymphoma, sepsis, and intracranial haemorrhage (one each) rather than directly of severe diarrhoea. Compared to the patients with the same PIDs without the SD phenotype (Table 3 and Supplemental Fig. 2), those with XLA and SCID with the SD phenotype trended to have a lower mortality rate. In contrast, the patients with WAS, CGD and HIGM with the SD phenotype had a relatively higher mortality rate, although none of the differences reached statistical significance except XLA patients.
In contrast to the SD group, the higher mortality rate in the RD group (two deaths of five patients; 40%) was directly related to their overwhelming RD phenotype. The parents of patient 1 (with an XIAP mutation) chose a conservative strategy rather than HSCT despite a poor response to aggressive treatment and methylprednisolone. Anti-TNF-α biologics were considered, however the RD phenotype worsened and endemic norovirus enterocolitis prevailed. Unfortunately, he died of severe Crohn’s colitis and accelerated hemophagocytic lymphohistiocytosis. Patient 3 (with a FOXP3 mutation) succumbed from recurrent RD while waiting for a suitable HSCT donor. Patient 2 (with a STAT1 mutation) underwent successful HSCT to terminate recurrent opportunistic and life-threatening infections as well as the RD phenotype. Patient 5 had broader gastrointestinal tract lesions from the stomach to the colon and lost 3 kg after having diarrhoea for 2 months. Concomitant Helicobacter pylori colonization was found in the second endoscopic stomach specimen, and additional amoxicillin and clarithromycin (2 weeks) and omeprazole (12 weeks) treatment attenuated the cramping and frequent diarrhoea.
Discussion
This is the first study to report the prevalence, distribution, pathogens and prognosis of SD from a national PIDs referral centre. Twenty-six patients (10.6%) who had insufficient opsonized and neutralized IgG (Btk, CVID, SCID and HIGM), diminished reactive oxygen species production (CGD), and lazy lymphocytes with cytoskeletal dysfunction (WAS), thus impairing pathogenic eradication developed the SD phenotype before adequate prophylactic therapy and needed an extended duration of antibiotic treatment. Severe bacterial colitis in immune-competent patients often responds to effective antibiotics within 7 days in Taiwan in the previous study42. Of note, pseudomonas colitis was most commonly found in the patients with predominantly antibody deficiencies, of whom one (Btk2) met the diagnostic criteria43 of Shanghai fever including community-acquired diarrhoea with fever, sepsis and pseudomonas cultures from facial ecthyma gangrenosum. His pathogen was not a multi-drug resistant strain.
In the pathogenesis of the extreme presentation of the RD phenotype, persistent intestinal inflammation seemed to play a greater role than infection, reflecting the poor response to antibiotics. Therefore, alternative biologics or anti-inflammatory immuno-suppressants stronger than steroids should be administered to patients with XIAP, STAT1, FOXP3, or STAT3 mutations. While alimentary enterocytes with these mutations interact with microorganisms for gut homeostasis, the XIAP mutation selectively impairs nucleotide-binding and oligomerization domain 1/2 signalling for chemo-attractant production (IL-6, IL-8, and MCP-1), and does not effectively terminate cognate caspase-related cascades for apoptosis in damaged and/or infected intestinal epithelium32,33,34,35. The dominant gain-of-function hypermorphic [Thr385Met] STAT1 mutation prolongs STAT1 phosphorylation to exaggerate IFN-γ, IFN-α, IL-6, and IL-21 cytokine signaling39,40,41. The FOXP3 mutation in Treg cells attenuates IL-10 and TGF-β production to release unwanted autoantibodies in autoimmune disorders17,18,19,20,21. In addition, we previously showed that the STAT3 mutation diminishes Th17 production44, inhibiting the intestinal tight junction45, 46, epithelial cell proliferation, goblet cell restoration, and mucin production47, 48. Furthermore, a novel suppressive Th17 subset has been shown to cause regulatory Th17 (rTh17) cells to induce mucosal tolerance when Th17 cells are recruited to the intestinal tract49. Taken together, the pathologic mechanisms orchestrated by these mutations break intestinal mucosal tolerance and augment persistent intestinal inflammation, thereby causing the RD phenotype. Successful HSCT is, in practice, rescue therapy to terminate the RD phenotype in patients with XIAP, FOXP3 and STAT1 mutations as well as recurrent infections50.
Helicobacter pylori-infected gastric mucosa can express higher levels of IL-17 via the STAT3 pathway51 and promote intestinal metaplasia and dysplasia to carcinoma in PIDs patients in whom malignant transformation has been reported to be 10 times higher than the general population52. However, the lack of IL-17 secretion in patients with the loss-of-function STAT3 mutation due to defective Th17 cell development does not enhance alimentary malignant transformation to induce the tumour-secreting-endocrine RD phenotype. Whether the Helicobacter pylori infection in our patient 5 with a STAT3 mutation was related to the RD phenotype is unclear, and further large-scale prospective studies are warranted to elucidate this issue.
There are several limitations to this study. First, some aetiological pathogens and triggering factors of SD were not identified, even though comprehensive PCR and stool cultures were performed to detect viruses, fungi, parasites and bacteria. Clearly delineating between infection and inflammation remains a challenge in PIDs patients with immune disturbances. Hence, both empiric antibiotics and steroids should be recommended to those with the RD phenotype. Second, in three RD patients with FOXP3, XIAP, and STAT1 mutations, successful HSCT reconstructed immunity and therefore resolved the RD phenotype as well as the recurrent infections. However, only one patient with the STAT1 mutation received timely HSCT. The hesitation of the other two parents to undergo HSCT increased the mortality rate despite an early diagnosis and aggressive treatment. Third, the prevalence of IBD in PIDs patients is under-estimated. IBD or IBD-like diarrhoea may emerge at an older age since the life expectancy of PIDs patients has increased due to advanced treatment strategies, thereby increasing the potential to develop inflammation and autoimmune disorders. The limited follow-up duration of our patients with predominantly paediatric-onset PIDs may explain the lower prevalence of IBD or IBD-like diarrhoea.
In conclusion, the 26 (10.5%) patients in this nationwide cohort of 246 PIDs patients were mostly male and developed the SD phenotype before adequate prophylactic treatment. Most cases were caused by salmonella and pseudomonas infections, and they needed IVIG and an extended period of antibiotic treatment. Five (2.0%) male patients with the RD phenotype had XIAP mutations resulting in defective nucleotide-binding and oligomerization domain 1/2 signalling, the hypermorphic STAT1 mutation causing hyper-cytokine signalling, and profound Treg and Th17 cell defects caused by FOXP3 and STAT3 mutations. Early recognition and timely effective interventions to suppress inflammation are beneficial to decrease the mortality rate of PIDs patients with the RD phenotype.
Methods
Patients
From 2003 to 2015, 246 index cases with PIDs from 215 families were identified from the PICAR Institute’s registry (Supplemental Table 1)37. Infectious colitis was diagnosed if the clinical manifestations of bloody diarrhoea, tenesmus, severe cramping abdominal pain and a high CRP level (>40 mg/mL) were unequivocal even if no pathogen was isolated after extensive culture and PCR amplification. According to our previous studies on the epidemiologic intestinal pathogens in patients with PIDs37, 53, ceftriaxone (100 mg/kg/day) or meropenem (30 mg/kg/day) was given for suspected Salmonella or Shigella colitis, and a combination of ceftazidime (100 mg/kg/day) and amikacin (15 mg/kg/day) or carbapenem (100 mg/kg/day) for pseudomonas colitis in the presence of neutropenia resembling Shanghai fever was given within the first 24 hours before the pathogen cultures were available43. When the blood cultures were sterile and infectious manifestations of tenesmus, severe cramping abdominal pain, fever, and elevated CRP level subsided, the antibiotics were discontinued.
Regardless of whether empiric or sensitive antibiotics were given for the identified pathogens and IVIG (0.6–0.8 g/kg) for the patients with hypogammaglobulinemia, frequent diarrhoea of more than three episodes of unformed stools within a period of 24 hours for longer than 2 weeks was defined as the SD phenotype. In cases of frequent diarrhoea that failed to respond to antibiotics, one dose of IVIG and steroids (methylprednisolone 2–5 mg/kg/day) for more than 3 weeks was defined as the refractory SD (RD) phenotype in this study. The prognosis of the patients with PIDs suffering from SD and RD were compared using Kalpan-Meier survival (GraphPad software) tests. For those without SD, the starting time of K-M analysis was the diagnosis day by characteristic presentations or/and molecular and genetic confirmation. They had no the SD phenotype to date. For those with SD, the starting time of K-M analysis was the diagnosis day by characteristic presentations accompanying the SD phenotype or/and molecular and genetic confirmation.
The Chang Gung Human Investigation Committee approved all carried methods and all experimental protocols in this study, and the patients’ parents or guardians’ informed consent were obtained. We confirmed that all methods were performed in accordance with the relevant guidelines and regulations.
Immunologic Functional Assessment and Candidate Gene Approach
In addition to immunoglobulin levels and lymphocyte subpopulations (i.e., CD3+, CD4+, CD4 + CD45RA+, CD4 + CD45RO+, CD19+, CD19 + CD27+, and CD19 + CD27−), characteristics and well-syndromes of PIDs accompanying the SD phenotype were identified by experienced physicians, and were consistent with defective lymphocyte proliferation to mitogens and antigens, and phorbol myristate acetate-stimulated polymorphonuclear reactive oxygen species as represented by H2O2 production as previously described54.
To evaluate IL-10 signalling in the patients with RD and unrecognized molecular defects at that time, purified PBMCs (5 × 106/ml) were stimulated overnight with 50 ng/ml E. coli LPS (Sigma-Aldrich, St. Louis, Mo) alone or with 20 ng/ml recombinant human IL-10 (R&D Systems, Minneapolis, MN). The supernatants were analysed using a commercially available TNF-α ELISA development kit in duplicate using a Tecan Sunrise ELISA micro-plate reader (R&D Systems, Minneapolis, MN). Candidate genetic analysis in the patients with the RD phenotype predisposing to IBD included, at least, IL10, IL10RA, IL10RB, NEMO, FOXP3, XIAP, STAT3 and STAT155, 56. Every two oligonucleotide primers were selected to cover the entire coding region by Sanger sequencing57.
References
Agarwal, S. & Mayer, L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin. Gastroenterol. Hepatol. 11, 1050–1063 (2013).
Agarwal, S. & Mayer, L. Gastrointestinal manifestations in primary immune disorders. Inflamm. Bowel Dis. 16, 703–711 (2010).
Agarwal, S. & Mayer, L. Pathogenesis and treatment of gastrointestinal disease in antibody deficiency syndromes. J. Allergy Clin. Immunol. 124, 658–664 (2009).
Mak, T. W. & Saunders, M. E. B cell development, activation and effector functions. In Primer to the Immune Response. MA, CA: 94–95 (Elsevier, 2011).
Catassi, C., Fabiani, E., Spagnuolo, M. I., Barera, G. & Guarino, A. Severe and protracted diarrhea: results of the 3-year SIGEP multicenter survey. Working Group of the Italian Society of Pediatric Gastroenterology and Hepatology (SIGEP). J. Pediatr. Gastroenterol. Nutr. 29, 63–68 (1999).
Guarino, A. et al. Etiology and risk factors of severe and protracted diarrhea. J. Pediatr. Gastroenterol. Nutr. 20, 173–178 (1995).
Uhlig, H. H. et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 147, 990–1007 (2014).
Uhlig, H. H. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 62, 1795–1805 (2013).
Jeffrey Modell Foundation. http://www.info4pi.org/newsletters website.
Li, Y. et al. New insights into the role of STAT3 in IBD. Inflamm. Bowel Dis. 18, 1177–183 (2012).
Lees, C. W. et al. New IBD genetics: common pathways with other diseases. Gut. 60, 1739–1753 (2011).
Cho, J. H. & Brant, S. R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 140, 1704–1712 (2011).
Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 58, 1152–1167 (2009).
Kobayashi, T. et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 57, 1682–1689 (2008).
Amre, D. K. et al. Association between genetic variants in the IL-23R gene and early-onset Crohn’s disease: results from a case-control and family-based study among Canadian children. Am. J. Gastroenterol 103, 615–620 (2008).
Duerr, R. H. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 314, 1461–1463 (2006).
Glocker, E. O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009).
Glocker, E. O. et al. Infant colitis - it’s in the genes. Lancet. 376, 1272 (2010).
Kotlarz, D. et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease - implications for diagnosis and therapy. Gastroenterology. 143, 347–355 (2012).
Mao, H. et al. Exome sequencing identifies novel compound heterozygous mutations of IL-10 receptor 1 in neo-natal onset Crohn’s disease. Genes Immun. 13, 437–442 (2012).
Engelhardt, K. R. et al. Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 131, 825–830 (2013).
Alangari, A. et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J. Allergy Clin. Immunol. 130, 481–488 (2012).
Cannioto, Z. et al. IBD and IBD mimicking enterocolitis in children younger than 2 years of age. Eur. J. Pediatr. 168, 149–155 (2009).
Ozgür, T. T. et al. Hematopoietic stem cell transplantation in a CD3 gamma-deficient infant with inflammatory bowel disease. Pediatr. Transplant. 12, 910–913 (2008).
Torgerson, T. R. & Ochs, H. D. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J. Allergy Clin. Immunol. 120, 744–750 (2007).
Barzaghi, F., Passerini, L. & Bacchetta, R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol 3, 211 (2012).
Catucci, M. et al. Autoimmunity in Wiskott-Aldrich syndrome: an unsolved enigma. Front Immunol 3, 209 (2012).
Dupuis-Girod, S. et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 111, e622–627 (2003).
Cannioto, Z. et al. IBD and IBD mimicking enterocolitis in children younger than 2 years of age. Eur. J. Pediatr. 168, 149–155 (2009).
Marks, D. J. et al. Inflammatory bowel disease in CGD reproduces the clinico-pathological features of Crohn’s disease. Am. J. Gastroenterol. 104, 117–124 (2009).
Muise, A. M. et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 61, 1028–1035 (2012).
Marsh, R. A. et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lympho-histiocytosis and not as X-linked lympho-proliferative disease. Blood. 116, 1079–1082 (2010).
Pachlopnik Schmid, J. et al. Clinical similarities and differences of patients with X-linked lympho-proliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 117, 1522–1529 (2011).
Yang, X. et al. Clinical and genetic characteristics of XIAP deficiency in Japan. J. Clin. Immunol. 32, 411–420 (2012).
Speckmann, C. et al. X-linked inhibitor of apoptosis (XIAP) deficiency: The spectrum of presenting manifestations beyond hemophagocytic lympho-histiocytosis. Clin. Immunol. 149, 133–141 (2013).
Cheng, L. E. et al. Persistent systemic inflammation and atypical enterocolitis in patients with NEMO syndrome. Clin. Immunol. 132, 124–31 (2009).
Lee, W. I. et al. Distribution, clinical features and treatment in Taiwanese patients with symptomatic primary immunodeficiency diseases (PIDs) in a nationwide population-based study during 1985–2010. Immunobiology. 216, 1286–1294 (2011).
Picard, C. et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J. Clin. Immunol. 35, 696–726 (2015).
Uzel, G. et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune-dysregulation-polyendocrinopathy- enteropathy-X-linked-like syndrome. J. Allergy Clin. Immunol. 13, 1611–1623 (2013).
Verbsky, J. W. & Chatila, T. A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr. Opin. Pediatr. 25, 708–714 (2013).
Sampaio, E. P. et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J. Allergy Clin. Immunol. 131, 1624–1634 (2013).
Chen, H. M., Wang, Y., Su, L. H. & Chiu, C. H. Nontyphoid salmonella infection: microbiology, clinical features, and antimicrobial therapy. Pediatr. Neonatol. 54, 147–152 (2013).
Chuang, C. H. et al. Shanghai fever: a distinct Pseudomonas aeruginosa enteric disease. Gut. 63, 736–743 (2014).
Lee, W. I. et al. Clinical, immunological and genetic features in Taiwanese patients with the phenotype of hyper-immunoglobulin E recurrent infection syndromes (HIES). Immunobiology. 216, 909–917 (2011).
Kinugasa, T. et al. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology. 118, 1001–1011 (2000).
Chen, Y. et al. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem. 278, 17036–17043 (2003).
Brand, S. et al. IL-22-mediated liver cell regeneration is abrogated by SOCS-1/3 over-expression in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1019–1028 (2007).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Esplugues, E. et al. Control of TH17 cells occurs in the small intestine. Nature. 475, 514–518 (2011).
Slatter, M. A. & Gennery, A. R. Advances in hematopoietic stem cell transplantation for primary immuno-deficiency. Expert Rev. Clin. Immunol. 9, 991–999 (2013).
Caruso, R. et al. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur. J. Immunol. 38, 470–478 (2008).
Dhalla, F., da Silva, S. P., Lucas, M., Travis, S. & Chapel, H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin. Exp. Immunol 165, 1–7 (2011).
Lee, W. I. et al. Distribution, infections, treatments and molecular analysis in a large cohort of patients with primary immunodeficiency diseases (PIDs) in Taiwan. J. Clin. Immunol. 26, 274–283 (2006).
Lee, W. I. et al. Distribution and clinical aspects of primary immunodeficiencies in a Taiwan pediatric tertiary hospital during a 20-year period. J. Clin. Immunol. 25, 162–173 (2005).
Lee, W. I. et al. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 105, 1881–1890 (2005).
Lee, W. I. et al. Clinical aspects and genetic analysis of Taiwanese patients with the phenotype of hyper-immunoglobulin E recurrent infection syndromes (HIES). J. Clin. Immunol. 31, 272–280 (2011).
RAPID: Resource of Primary Immunodeficiency Diseases. http://rapid.rcai.riken.jp/ website.
Acknowledgements
The authors thank all of the patients and their families for their kind cooperation and their physicians for the referrals. This study received grants from Chang-Gung Medical Research Progress (Grant CORPG3F0051-53 and CMRPG 4B0051-53), National Science Council (Grants NSC 99-2314-B-182A-096-MY3, NSC 102-2314-B-182A-039-MY3, MOST 104-2663-B-182A-005 and NMRPG3C6071) and Taiwan Foundation for Rare Disorders (TFRD).
Author information
Authors and Affiliations
Contributions
L.W.I., C.C.C. and H.J.L. designed the study and organised the team; L.W.I., H.J.L., and O.L.S. performed the immunologic assessments and genetic analysis; C.C.C., J.T.H. and C.C.C. took care of the patients; and H.C. studied the pathology. All of the authors contributed to this work and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, WI., Chen, CC., Jaing, TH. et al. A Nationwide Study of Severe and Protracted Diarrhoea in Patients with Primary Immunodeficiency Diseases. Sci Rep 7, 3669 (2017). https://doi.org/10.1038/s41598-017-03967-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03967-4
This article is cited by
-
Clinical Features and Genetic Analysis of Taiwanese Primary Immunodeficiency Patients with Prolonged Diarrhea and Monogenetic Inflammatory Bowel Disease
Journal of Clinical Immunology (2023)
-
Chronic Diarrhoea in Infants and Children: Approaching and Managing the Problem
Current Treatment Options in Pediatrics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
