
Abstract
Syndromic primary immunodeficiencies are rare genetic disorders that affect both the immune system and other organ systems. More often, the immune defect is not the major clinical problem and is sometimes only recognized after a diagnosis has been made based on extra-immunological abnormalities. Here, we report two sibling pairs with syndromic primary immunodeficiencies that exceptionally presented with a phenotype resembling early-onset common variable immunodeficiency, while extra-immunological characteristics were not apparent at that time. Additional features not typically associated with common variable immunodeficiency were diagnosed only later, including skeletal and organ anomalies and mild facial dysmorphism. Whole exome sequencing revealed KMT2A-associated Wiedemann-Steiner syndrome in one sibling pair and their mother. In the other sibling pair, targeted testing of the known disease gene for Roifman syndrome (RNU4ATAC) provided a definite diagnosis. With this study, we underline the importance of an early-stage and thorough genetic assessment in paediatric patients with a common variable immunodeficiency phenotype, to establish a conclusive diagnosis and guide patient management. In addition, this study extends the mutational and immunophenotypical spectrum of Wiedemann-Steiner and Roifman syndromes and highlights potential directions for future pathophysiological research.
Similar content being viewed by others

Introduction
Common variable immunodeficiency (CVID) is one of the most frequently diagnosed primary immunodeficiencies (PIDs), and is defined as decreased serum immunoglobulin (Ig) G, decreased IgA and/or IgM, poor antibody responses to vaccines, and exclusion of other causes of hypogammaglobulinemia1. Patients commonly experience recurrent (sinopulmonary) infections and features of immune dysregulation such as autoimmunity1, 2. About 25% of CVID patients are diagnosed in childhood3. To rule out transient hypogammaglobulinemia of infancy, in which Ig levels spontaneously resolve mostly by the age of two to four years, a definite diagnosis of CVID should not be given before the age of four years1.
Here, we report novel familial cases of Wiedemann-Steiner syndrome (WSS) and Roifman syndrome (RS) that were initially categorized as early-onset CVID. WSS and RS are rare syndromic PIDs, affecting the immune system as well as other organ systems4,5,6. Although there is considerable phenotypic heterogeneity in both syndromes, hallmark extra-immunological features are generally evident very early in life7, 8. WSS is typically characterized by hypertrichosis cubiti, growth retardation, developmental delay and facial dysmorphism, and is caused by heterozygous mutations in lysine methyltransferase 2 A (KMT2A)7, 9,10,11,12,13,14. KMT2A (also called mixed-lineage leukemia, MLL) encodes a histone methyltransferase involved in regulating chromatin-mediated transcription and is a frequent target of chromosomal rearrangements in childhood leukemia9, 15. WSS has only been recently associated with primary antibody deficiency7. RS, on the other hand, is commonly featured by antibody deficiency as well as growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy8, 16. Biallelic mutations in RNU4ATAC, a noncoding small nuclear RNA (snRNA) gene, were recently identified as a cause of RS8. U4atac snRNA is an important component of the minor spliceosome required for minor intron splicing8.
This report aspires to increase awareness among immunologists and geneticists that a CVID phenotype can be the principal presentation of WSS and RS in early childhood, which is exceptional and has not been previously reported. The prior diagnosis of early-onset CVID diverted attention away from the initially less evident extra-immunological features, which significantly delayed identification of the underlying syndromic disorders. Additionally, in both families we identified mutations that have not been previously associated with disease. We aimed to provide insight as to how these mutations are disease-causing. Finally, we expand the immunophenotypical spectrum of WSS and RS, which could support future mechanistic research.
Results
An early-onset CVID phenotype in two unrelated sibling pairs
This study reports on two unrelated sibling pairs with recurrent respiratory tract infections and antibody deficiency categorized as CVID in early childhood. The family A monozygotic twin boys (Fig. 1a, II:2 and II:3) were born prematurely at 34 weeks gestational age to non-consanguineous, Belgian parents and are currently 11 years old. One of them (II:3) was born with bilateral inguinal hernia and hypospadias, which were attributed to his premature birth. From the first year of life, the twin boys suffered from recurrent upper and lower respiratory tract infections, often requiring antibiotics. At 2 months of age, patient II:3 developed severe pneumonia with respiratory arrest and heart failure. The latter led to the recognition of a patent ductus arteriosus, which was surgically ligated shortly thereafter. The postoperative course was complicated by severe respiratory distress requiring ventilation and systemic corticosteroids. Upon immunological evaluation, both patients II:2 and II:3 demonstrated panhypogammaglobulinemia, poor antibody responses to Pneumococcal polysaccharide vaccine, increased naive B cells, and very low memory B cells (Table 1). Additionally, both patients showed evidence of mild bronchiectasis on high-resolution computed tomography (HRCT) scan at 3.5 years of age.

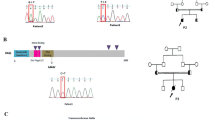
Family A with KMT2A-associated Wiedemann-Steiner syndrome (WSS). (a) Pedigree of family A. (b) Skipping of KMT2A exon 28. Gel electrophoresis of the KMT2A cDNA region containing exon 28 revealed a second shorter transcript in the three affected individuals. HC1 and HC2 represent two healthy controls; GAPDH was used as reference target. In-frame deletion of exon 28 was confirmed by cDNA sequencing; c.10755 and c.10835 indicate the start respectively stop position of exon 28. (c) KMT2A protein domains (adapted from ref. 9). KMT2A is cleaved in an N-terminal (KMT2A-N) and C-terminal (KMT2A-C) fragment, which form a non-covalently associated complex. Deletion of the amino acids encoded by exon 28 may disrupt the interaction site between the two fragments.
The family B brother and sister pair (Fig. 2a, II:1 and II:2) were born to healthy, non-consanguineous, Belgian parents and are currently 17 and 14 years of age respectively. They had recurrent upper and lower respiratory tract infections since the age of 18 months (II:1) and 3 years (II:2). The girl (II:2) also had recurrent gastroenteritis in early childhood and diffuse atopic eczema since infancy. Similar to the family A sibling pair, they had panhypogammaglobulinemia and poor global antibody responses to Pneumococcal polysaccharide vaccine (Table 2). Serotype-specific Pneumococcal antibody responses were not evaluated. In contrast to the family A twins, the family B siblings demonstrated severe B cell lymphopenia with normal switched memory B cell percentages and increased CD21low B cell levels (Table 2). HRCT scan in patient II:1 at the age of 7.5 years displayed marked bronchiectasis, whereas his sister (II:2) only showed discrete bronchiectasis at a similar age.

Family B with RNU4ATAC-associated Roifman syndrome (RS). (a) Pedigree of family B. (b) Representative retinal images of the RS patients. Panel I, composite retinal image of fundus of left eye (LE) of patient II:1: note inferior outer retinal atrophy with greyish hue and intraretinal pigment migration of the spicular type in inferior retina; mottled aspect of retinal pigment epithelium, more pronounced in inferotemporal area. Panel II, blue light autofluorescence image of LE of patient II:1 showing hyperautofluorescent delineation of inferior atrophic zone, as well as superior to optic disc, illustrating more widespread disease than can be seen on white light fundoscopic image only. Panel III, similar image of right eye (RE) of patient II:1 as in Panel II. Panel IV, fundus picture of detail of superonasal midperiphery of RE of patient II:2. Despite a normal full-field flash electroretinography, recent fundus examination at 14 years of age showed a mild mottling of pigment epithelium suggestive of early stage retinal dystrophy. (c) U4atac snRNA showing structural elements, conserved positions and location of variants associated with RS (adapted from ref. 8). The here-reported variant that has not been previously associated with RS is shown in red.
In both the family A and B sibling pairs, the clinical presentation and laboratory findings in the first years of life were reminiscent of a CVID phenotype. All patients are currently doing well under regular Ig replacement therapy, antiflogistic maintenance treatment with azithromycin and intermittent inhaled corticosteroids and/or short-acting beta-agonists therapy.
Extra-immunological features raised suspicion of syndromic PID
Although both sibling pairs first presented with a phenotype resembling CVID, with time they gradually demonstrated additional clinical features not typically associated with CVID (Tables 3 and 4). The family A twin boys developed a third degree atrioventricular block for which a pacemaker was implanted at 5 (II:3) and 6.5 (II:2) years of age respectively. In the following years, the boys showed increasing evidence of mild intellectual disability. In retrospect, patient II:3 had signs of mild developmental delay during the first years of life. Both twins also demonstrated poor weight gain and growth retardation, albeit to a limited extent. Around the age of 9 years, dysmorphic facial features became more conspicuous (Table 3). Interestingly, the twins’ mother (I:2) had congenital urogenital tract anomalies consisting of a unicornuate uterus and a unique left ovary, fallopian tube and kidney. Moreover, since childhood, she had suffered from right unilateral sensorineural hearing loss as well as recurrent sinusitis and bronchitis frequently requiring antibiotics. At that time, genetic or immunological testing had never been performed in the mother as she deemed herself to be in good general health. The twins’ older brother (II:1) and father (I:1) had an uneventful medical history.
Analogously, the boy in family B (II:1) initially displayed subtle syndromic features, such as mild growth retardation, that appeared more pronounced over time. At the age of seven years, diverse skeletal abnormalities including spondyloepiphyseal dysplasia were detected (Table 4). At the same age, he was also found to have slowly progressive retinal dystrophy (Fig. 2b, Panels I-III) with decreased rod function but near-normal cone function on full-field flash electroretinography. Antibody deficiency in combination with skeletal and ophthalmological features led to the clinical suspicion of RS in the boy. However, his sister (II:2) had no radiographic sings of spondyloepiphyseal or hip dysplasia nor retinal dystrophy. Moreover, RS was originally presumed to be an X-linked recessive condition, although no causal gene had been identified16. When the family B girl (II:2) was about 9 years old, Gray et al. reported the first female patient with RS, having a skewed X-inactivation and a milder phenotype than her affected brother17. Subsequently, we hypothesized that patient II:2 might be a manifesting heterozygote of RS, which could be compatible with her milder extra-immunological phenotype at that time.
Cytogenetic and cytogenomic analyses were negative in both sibling pairs
In the family A twins, conventional G-banding karyotype, fluorescent in situ hybridization for region 22q11.2 and subtelomeric screening were normal. Furthermore, microarray-based comparative genomic hybridisation analysis in both sibling pairs did not demonstrate copy number variations.
Whole exome sequencing (WES) uncovers WSS in family A
Since no specific genetic syndrome was suspected in family A, WES was performed in patient II:2 and both parents. This revealed a heterozygous splice site variant in KMT2A (NM_001197104:c.10835 + 1 G > A), present in the twins (II:2, II:3) as well as in the mother (I:2) (Fig. 1a). The variant is not reported in public or in-house databases. The KMT2A nucleotide substitution is situated in the splice donor site of intron 28. In silico splicing prediction tools suggested complete loss of the splice donor site resulting in exon 28 skipping and an in-frame deletion of 81 bp, which was confirmed by analyses on cDNA derived from patients’ PBMCs (Fig. 1b). Mature KMT2A protein is physiologically cleaved in an N-terminal (KMT2A-N) and C-terminal (KMT2A-C) fragment, which together form a non-covalently associated complex (Fig. 1c)15, 18. Complex formation is necessary for stability and subnuclear localization of the protein18. The amino acids encoded by exon 28 are part of the interaction site between KMT2A-N and KMT2A-C (Fig. 1c)15. It has been shown that disrupting the interaction between the two fragments causes degradation of the KMT2A-N fragment and loss of protein function18. The KMT2A-N fragment was only very weakly detectable by western blot on PBMC lysates, however, and could therefore not be reliably interpreted (data not shown). Subsequent investigations in the mother demonstrated mild intellectual disability, undetectable serum IgM, and reduced switched memory B cells (Table 1). Taken together, the c.10835 + 1 G > A variant in KMT2A indicates a molecular diagnosis of WSS in the twin brothers and their mother.
Targeted sequencing confirms the diagnosis of RS in family B
In family B, WES was unable to identify a potentially disease-causing variant. In 2015, biallelic mutations in RNU4ATAC were identified in patients with RS8. Since RNU4ATAC is a noncoding snRNA gene, possible variants would have been missed with WES. Indeed, subsequent Sanger sequencing of RNU4ATAC revealed compound heterozygous variants in both siblings (c.13 C > T and c.116 A > T) that segregated in the parents (Fig. 2a). The c.13 C > T variant (rs559979281) had been previously reported in RS (Fig. 2c)8. The c.116 A > T variant has, to our knowledge, not yet been associated with human disease. The public database gnomAD (Genome Aggregation Database) contains two heterozygotes for the c.116 A > T variant (allele frequency of 0.00001531) but no homozygotes. Importantly, the c.116 A > T variant is located in a highly conserved position involved in splicing activity (Fig. 2c)8. Furthermore, position 116 is immediately adjacent to the Sm protein-binding site, which is a highly conserved structural element essential in splicing activity and previously implicated in RS (Fig. 2c)8. Together, the RNU4ATAC genotype confirms the diagnosis of RS in the family B siblings.
Immunological abnormalities in the WSS and RS patients
Because of the prominent immunodeficiency in both sibling pairs, we performed flow cytometric analysis of B and T lymphocyte subsets as previously described19. Interestingly, all patients from families A and B had decreased circulating follicular helper T (cTfh) cells (Fig. 3a,b). Tfh cells play an essential role in the formation of antibody-producing plasma cells and memory B cells20. Furthermore, the two RS patients showed markedly reduced expression levels of B cell activating factor-receptor (BAFF-R), a receptor important in peripheral B cell survival (Fig. 3b)21. The WSS patients had normal BAFF-R levels (Fig. 3a). Expression of transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), a receptor related to BAFF-R, was normal in both the WSS and RS patients (Fig. 3a,b)21. For all patients, the alterations in naive and memory lymphocyte subsets (Supplementary Figs 1–4) corresponded with those seen in the routine laboratory assessment (Table 1). Other examined B and T cell populations fell within the range of the age-matched healthy controls (Supplementary Figs 1–4).

cTfh cells, BAFF-R expression and TACI expression in WSS and RS patients. (a) Family A patients with KMT2A-associated Wiedemann-Steiner syndrome (WSS). The twins (II:2, II:3) were 8 years old and the mother (I:2) was 43 years old at time of analysis. (b) Family B patients with RNU4ATAC-associated Roifman syndrome (RS). At time of analysis, the patients (II:1, II:2) were 14 and 11 years old, respectively. Flow cytometric immunophenotyping was performed on patients’ PBMCs in comparison with age-matched healthy controls (HC). T cells were gated as CD3+ and B cells as CD19+CD20+ in total PBMCs. Circulating follicular helper T (cTfh) cells were gated as CXCR5+CD45RO+ in CD4+ T cells. BAFF-R and TACI expression were measured on B cells. Relative mean fluorescence intensity (MFI) was calculated by dividing the MFI of the positive population by the MFI of the Fluorescence Minus One (FMO) population. Graphs of the HC groups represent mean ± standard deviation. BAFF-R: B cell activating factor-receptor, cTfh: circulating follicular helper T, expr: expression, TACI: transmembrane activator and calcium modulator and cyclophilin ligand interactor.
Discussion
We report two sibling pairs with an early-onset CVID phenotype as primary and cardinal presentation of WSS and RS: recurrent sinopulmonary infections, panhypogammaglobulinemia, reduced polysaccharide vaccine responses, and aberrant peripheral B cell subsets1. Because extra-immunological features were initially subtle and only became conspicuous with age, the establishment of an accurate diagnosis was significantly delayed. Therefore, we recommend to proactively evaluate all paediatric patients with a CVID phenotype for extra-immunological syndromic features, especially when presenting at an early age. In particular, diagnostic workup should include evaluation by a clinical geneticist, in addition to orthopedic, cardiologic, neurologic, urogenital and ophthalmologic assessment. To reach a conclusive diagnosis, genetic testing should be performed, varying from targeted testing of a specific disease gene to WES. Here, the diagnosis of WSS in family A was only confirmed upon WES in affected family members7, 9. In family B on the other hand, WES failed to reveal the causal genetic defect because this was located in a noncoding gene8. Targeted testing of the known disease gene for RS allowed to identify the underlying mutations and to provide a definite diagnosis8. Of note, if WES does not identify a genetic defect and there is no known disease gene, whole genome sequencing should be undertaken22.
With this study, we extend the phenotypical and mutational spectrum of both KMT2A-associated WSS and RNU4ATAC-associated RS7,8,9,10,11,12,13,14. In family A, we identified a novel heterozygous splice site mutation in KMT2A causing in-frame deletion of exon 28. This deletion likely disrupts the stabilizing interaction site between the N- and C-terminal KMT2A fragments, resulting in loss of protein function18. So far, we were unable to confirm this on a protein level because the KMT2A-N protein fragment was not reliably detectable by western blot. Further studies on protein level are planned in the future. Family A is the first published kindred to show autosomal dominant transmission of KMT2A-associated WSS in multiple generations, as previously reported cases were either sporadic or parents were unavailable7. Remarkably, the characteristic hypertrichosis of elbows, back and/or lower limbs was absent in the here-reported WSS patients, confirming previous literature that this feature may not be as pathognomonic as initially believed7, 9, 11, 12. Interestingly, KMT2A-associated WSS shows phenotypical overlap with Kabuki syndrome type 1 caused by heterozygous mutations in the related gene KMT2D 23. Over 80% of patients with KMT2D-associated Kabuki syndrome develop defects in terminal B cell differentiation resulting in antibody deficiency23. Similarly, the here-described WSS patients demonstrated a block in terminal B cell differentiation evidenced by a relative increase in transitional and naive B cells and a relative decrease in switched memory B cells. Moreover, they had reduced levels of cTfh cells, which play a pivotal role in terminal B cell differentiation20. Decreased cTfh cells have, to our knowledge, not been previously reported in WSS or Kabuki syndrome. It would be interesting to investigate cTfh cells in additional patients with KMT2A-associated WSS and KMT2D-associated Kabuki syndrome as this may help elucidate the underlying pathophysiology of the antibody deficiency. In summary, humoral immune deficiency in patients with WSS reported by us and by Stellacci et al.7, and antibody deficiency in patients with heterozygous mutations in the related KMT2D gene23, strongly suggest a previously unknown role for KMT2A in B cell biology that may be related with T helper cell function.
In family B, we identified rare compound heterozygous mutations in the noncoding RNU4ATAC gene, of which one mutation (c.116 A > T) had not been associated with RS before8. Although it was not initially apparent, with time the boy showed typical features of RS including spondyloepiphyseal dysplasia and retinal dystrophy8. Curiously, he also demonstrated bilateral agenesis of the anterior cruciate ligaments and the 12th ribs, which are not typically seen in RS8. Spondyloepiphyseal or hip dysplasia have not yet been documented in the girl, currently 14 years old, although she displays mild growth retardation. Only recently, she was found to have mild fundus abnormalities suggestive of early stage retinal dystrophy. Note that retinal dystrophy was already evident in her brother at 7 years of age. It would be interesting to investigate why the girl has a milder phenotype than her brother. Since U4atac snRNA plays a role in minor intron splicing, it would be valuable to conduct RNA sequencing analysis in the two siblings and check for possible differences in intron retention8. Detailed immunological workup in the RS patients revealed markedly decreased BAFF-R expression on B cells. To our knowledge, this finding has not been previously published. As BAFF-R signalling is important for survival of B cells in the peripheral blood, this may provide an important clue towards the B cell lymphopenia seen in RS patients21. Moreover, analogous to the WSS patients, the RS siblings demonstrated decreased levels of cTfh cells, which may further compromise B cell differentiation and antibody production20.
In conclusion, we here illustrate that a CVID phenotype can be the initial presentation of WSS and RS in early childhood while hallmark extra-immunological characteristics may be less prominent. With this, we highlight the importance of pursuing a genetic diagnosis in paediatric patients with an early-onset CVID phenotype, as this has important implications in terms of counselling, follow-up and screening for complications associated with the specific disorder.
Methods
Statement
All experiments and methods were carried out in accordance with relevant guidelines and regulations. The research protocol and all experimental protocols were approved by the ethical committee of Ghent University Hospital (2012/593). All reported subjects provided written informed consent for participation in the study, in accordance with the 1975 Helsinki Declaration.
Cytogenetic analyses
Microarray-based comparative genomic hybridization (array CGH) was performed on the affected sibling pairs of families A and B using the SurePrint G3 Human CGH Microarray Kit according to manufacturer’s instructions (Agilent Technologies). Results were analyzed with arrayCGHbase24. Karyotype analysis was performed on the family A twins using the conventional G-banding technique. To screen the family A twins for submicroscopic subtelomeric rearrangements, multiplex ligation-dependent probe amplification (MLPA) analysis was performed using SALSA P070 and SALSA P036C probe mixes according to manufacturer’s instructions (MRC-Holland). To examine the family A twins for 22q11.2 deletion, fluorescence in situ hybridization (FISH) analysis was performed using the DiGeorge Region Probe Set – LSI TUPLE 1 SpectrumOrange/LSI ARSA SpectrumGreen according to manufacturer’s instructions (Vyvis).
WES
Genomic DNA was isolated from whole blood leukocytes using the Puregene DNA isolation kit (Qiagen) according to manufacturer’s instructions. Whole exome enrichment was performed with the SureSelectXT Human All Exon V5 + UTRs kit (Agilent Technologies). Paired-end massively parallel sequencing (100 cycles) was performed on a NextSeq 500 (Illumina). Read mapping against the human genome reference sequence (NCBI, GRCh37), and post-mapping duplicate read removal, quality-based variant calling and coverage analysis were performed with CLC Genomics Workbench v6.0.4 (CLC bio). Sequencing coverage is summarized in Supplementary Table S1. Called variants with coverage ≥3 were annotated with Alamut Batch (Interactive Biosoftware). Only variants with population frequencies less than 10% were considered, according to public databases NCBI dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), ExAC Browser (http://exac.broadinstitute.org/), and 1000 Genomes Project Browser (http://browser.1000genomes.org/). Variants were further prioritized based on allele frequency, functional prediction scores, nucleotide conservation scores and biological relevance25. Both Mendelian and non-Mendelian inheritance patterns were taken into account. Afterwards, variants of interest were evaluated using Alamut Visual mutation interpretation software v2.7 rev. 1 (Interactive Biosoftware), Ingenuity Variant Analysis (QIAGEN, 2015 Release Spring), CADD scores v1.3 (http://cadd.gs.washington.edu/home), genome Aggregation Database (gnomAD) Browser (http://gnomad.broadinstitute.org), literature search, segregation analysis in available family members, and frequency in an in-house database containing variants of more than 1000 exomes at time of analysis.
Sanger sequencing of genomic DNA
DNA templates (GRCh37/hg19) of KMT2A (NM_001197104) and RNU4ATAC (NR_023343) were obtained from UCSC Genome Browser (https://genome.ucsc.edu). Primers for amplification and sequencing were designed with Primer3Plus26. For KMT2A exon 28 and adjacent intron-exon borders (family A): forward primer 5′-CAACCCACAAGGGTGTCTTC-3′ and reverse primer 5′-GCCCGGCTAATTCTTTTTGT-3′. For the unique exon and intron-exon borders of RNU4ATAC (family B): forward primer 5′-TGGAGGCTGGAGGTAAGCTA-3′ and reverse primer 5′-TGAGGTGCAAAGACCTACTGAA-3′. Genomic DNA was amplified by PCR using the specific primers and KAPA2G Robust Hotstart Ready Mix (KAPA Biosystems). PCR products were enzymatically purified with Exonuclease I and Antartic phosphatase (both New England BioLabs Inc.). Purified PCR products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) on a 3730xl DNA Analyzer (Applied Biosystems). Sequence reads were analyzed with SeqScape v2.5 (ThermoFisher Scientific).
RNA extraction, cDNA synthesis and confirmation of skipping of exon 28 in KMT2A
Total RNA was isolated from total PBMCs of all family A members and two control subjects by use of the RNeasy Plus Mini Kit (Qiagen) and reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad), according to manufacturer’s instructions. The cDNA template (GRCh37/hg19) of KMT2A (NM_001197104) was obtained from UCSC Genome Browser (https://genome.ucsc.edu). Primers for amplification and sequencing of exon 28 and adjacent coding regions were designed with Primer3Plus26: forward primer 5′-AACCCAAACCAAAAACCAAAC-3′ and reverse primer 5′-CATCAGTGGGGAGCTGAAAT-3′. GAPDH was used as a reference target: forward primer 5′-CAGCCTCAAGATCATCAGCA-3′ and reverse primer 5′-TGTGGTCATGAGTCCTTCCA-3′. PCR amplification was performed by use of GoTaq Hot Start Colorless Master Mix (Promega). PCR products were analyzed on a 2% agarose gel in 1x TBE buffer (Quality Biological Inc). SYBR Safe (Invitrogen) signals were captured with a Gel Doc EZ Imager system (Bio-Rad). In addition, purified PCR products were Sanger sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) on a 3130xL Genetic Analyzer (Applied Biosystems). Sequence reads were analyzed with SeqMan (DNAStar).
Flow cytometric analysis of PBMCs
Immunophenotyping was performed on PBMCs of patients and age-matched healthy controls. PBMCs were isolated from EDTA whole blood by Ficoll-Paque density gradient centrifugation and cryopreserved at −150 °C. Thawed PBMCs were stained with fixable viability dye 506 (eBioscience) and fluorescently labeled monoclonal antibodies under saturation conditions as previously described19. Cells were acquired on an LSR Fortessa flow cytometer (BD Biosciences). Data were analyzed with FlowJo version X (Tree Star Inc.).
References
Bonilla, F. A. et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract 4, 38–59, doi:10.1016/j.jaip.2015.07.025 (2016).
Bogaert, D. J. et al. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J. Med. Genet. 53, 575–590, doi:10.1136/jmedgenet-2015-103690 (2016).
Resnick, E. S., Moshier, E. L., Godbold, J. H. & Cunningham-Rundles, C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 119, 1650–1657, doi:10.1182/blood-2011-09-377945 (2012).
Ming, J. E., Stiehm, E. R. & Graham, J. M. Jr. Syndromic immunodeficiencies: genetic syndromes associated with immune abnormalities. Crit. Rev. Clin. Lab. Sci. 40, 587–642, doi:10.1080/714037692 (2003).
Kersseboom, R., Brooks, A. & Weemaes, C. Educational paper: syndromic forms of primary immunodeficiency. Eur. J. Pediatr. 170, 295–308, doi:10.1007/s00431-011-1396-7 (2011).
Ming, J. E. & Stiehm, E. R. Genetic syndromic immunodeficiencies with antibody defects. Immunol. Allergy Clin. North. Am. 28, 715–736, vii, doi:10.1016/j.iac.2008.06.007 (2008).
Stellacci, E. et al. Congenital immunodeficiency in an individual with Wiedemann-Steiner syndrome due to a novel missense mutation in KMT2A. Am. J. Med. Genet. A 170, 2389–2393, doi:10.1002/ajmg.a.37681 (2016).
Merico, D. et al. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun. 6, 8718, doi:10.1038/ncomms9718 (2015).
Jones, W. D. et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 91, 358–364, doi:10.1016/j.ajhg.2012.06.008 (2012).
Mendelsohn, B. A., Pronold, M., Long, R., Smaoui, N. & Slavotinek, A. M. Advanced bone age in a girl with Wiedemann-Steiner syndrome and an exonic deletion in KMT2A (MLL). Am. J. Hum. Genet. A 164a, 2079–2083, doi:10.1002/ajmg.a.36590 (2014).
Strom, S. P. et al. De Novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med. Genet. 15, 49, doi:10.1186/1471-2350-15-49 (2014).
Calvel, P. et al. A Case of Wiedemann-Steiner Syndrome Associated with a 46, XY Disorder of Sexual Development and Gonadal Dysgenesis. Sex. Dev. 9, 289–295, doi:10.1159/000441512 (2015).
Dunkerton, S. et al. A de novo Mutation in KMT2A (MLL) in monozygotic twins with Wiedemann-Steiner syndrome. Am. J. Med. Genet. A 167a, 2182–2187, doi:10.1002/ajmg.a.37130 (2015).
Miyake, N. et al. Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin. Genet. 89, 115–119, doi:10.1111/cge.12586 (2016).
Yokoyama, A., Kitabayashi, I., Ayton, P. M., Cleary, M. L. & Ohki, M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood 100, 3710–3718, doi:10.1182/blood-2002-04-1015 (2002).
Roifman, C. M. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: a novel syndrome. Clin. Genet. 55, 103–109, doi:10.1034/j.1399-0004.1999.550206.x (1999).
Gray, P. E., Sillence, D. & Kakakios, A. Is Roifman syndrome an X-linked ciliopathy with humoral immunodeficiency? Evidence from 2 new cases. Int. J. Immunogenet. 38, 501–505, doi:10.1111/j.1744-313X.2011.01041.x (2011).
Hsieh, J. J., Ernst, P., Erdjument-Bromage, H., Tempst, P. & Korsmeyer, S. J. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol. Cell. Biol. 23, 186–194, doi:10.1128/MCB.23.1.186-194.2003 (2003).
Bogaert, D. J. et al. The immunophenotypical fingerprint of patients with primary antibody deficiencies is partially present in their asymptomatic first-degree relatives. Haematologica 102, 192–202, doi:10.3324/haematol.2016.149112 (2017).
Liu, X., Nurieva, R. I. & Dong, C. Transcriptional regulation of follicular T-helper (Tfh) cells. Immunol. Rev. 252, 139–145, doi:10.1111/imr.12040 (2013).
Rickert, R. C., Jellusova, J. & Miletic, A. V. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immun. Rev 244, 115–133, doi:10.1111/j.1600-065X.2011.01067.x (2011).
Meienberg, J., Bruggmann, R., Oexle, K. & Matyas, G. Clinical sequencing: is WGS the better WES? Hum. Genet. 135, 359–362, doi:10.1007/s00439-015-1631-9 (2016).
Lindsley, A. W. et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J. Allergy Clin. Immunol. 137, 179–187.e110, doi:10.1016/j.jaci.2015.06.002 (2016).
Menten, B. et al. arrayCGHbase: an analysis platform for comparative genomic hybridization microarrays. BMC bioinformatics 6, 124, doi:10.1186/1471-2105-6-124 (2005).
Coppieters, F. et al. Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet. Med. 16, 671–680, doi:10.1038/gim.2014.24 (2014).
Untergasser, A. et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35, W71–74, doi:10.1093/nar/gkm306 (2007).
Shearer, W. T. et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J. Allergy Clin. Immunol. 112, 973–980, doi:10.1016/j.jaci.2003.07.003 (2003).
Piatosa, B. et al. B cell subsets in healthy children: reference values for evaluation of B cell maturation process in peripheral blood. Cytometry B Clin. Cytom 78, 372–381, doi:10.1002/cyto.b.20536 (2010).
Acknowledgements
The authors gratefully acknowledge the families who participated in this study. This study was supported by the Ghent University Hospital Spearhead Initiative for Immunology Research, the Jeffrey Modell Foundation to F.H., the Research Foundation Flanders (FWO) to D.B., B.P.L, F.C., B.N.L. and E.D.B., the Ghent University Special Research Fund (BOF15/GOA/011) to E.D.B., Hercules foundation AUGE/13/023 to E.D.B., a European Research Council consolidator grant to B.N.L., an Interuniversity Attraction Pole grant to B.N.L., the University of Ghent MRP program “Group-ID” to B.N.L., and the NIH Clinical Center intramural research program to H.S.K., J.E.N. and S.D.R. D.B. is a PhD fellow, F.C. a postdoctoral fellow, and E.D.B. and B.P.L. are Senior Clinical Investigators of the FWO.
Author information
Authors and Affiliations
Contributions
D.J.B. performed the genetic analyses, the experiments and data analysis, and drafted the initial manuscript. M.D., H.S.K. and S.D.R. supervised experiments and data analysis, and critically reviewed and revised the manuscript. J.E.N. assisted in protein structure analysis and critically reviewed and revised the manuscript. B.P.L., H.D.W., S.D.S., B.N.L. and F.D.B. managed patients, provided clinical data and critically reviewed and revised the manuscript. M.D.B. and F.C. assisted in genetic analyses and critically reviewed and revised the manuscript. E.D.B. supervised genetic analyses and critically reviewed and revised the manuscript. F.H. conceptualized the study, managed patients, provided and interpreted clinical data, and critically reviewed and revised the manuscript. All authors provided critical input and approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bogaert, D.J., Dullaers, M., Kuehn, H. et al. Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes. Sci Rep 7, 3702 (2017). https://doi.org/10.1038/s41598-017-02434-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02434-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
