
Abstract
The mitochondrial genome (mitogenome) can provide useful information for analyzing phylogeny and molecular evolution. Scorpaeniformes is one of the most diverse teleostean orders and has great commercial importance. To develop mitogenome data for this important group, we determined the complete mitogenomes of two gurnards Chelidonichthys kumu and Lepidotrigla microptera of Triglidae within Scorpaeniformes for the first time. The mitogenomes are 16,495 bp long in C. kumu and 16,610 bp long in L. microptera. Both the mitogenomes contain 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes and two non-coding regions. All PCGs are initiated by ATG codons, except for the cytochrome coxidase subunit 1 (cox1) gene. All of the tRNA genes could be folded into typical cloverleaf secondary structures, with the exception of tRNASer(AGN) lacks a dihydrouracil (DHU) stem. The control regions are both 838 bp and contain several features common to Scorpaeniformes. The phylogenetic relationships of 33 fish mitogenomes using Bayesian Inference (BI) and Maximum Likelihood (ML) based on nucleotide and amino acid sequences of 13 PCGs indicated that the mitogenome sequences could be useful in resolving higher-level relationship of Scorpaeniformes. The results may provide more insight into the mitogenome evolution of teleostean species.
Similar content being viewed by others


Introduction
Generally, the fish mitogenome is a circular and double-stranded molecule ranging from 15 to 19 kilobases in length. It usually contains two ribosomal RNA genes (12S rRNA and 16S rRNA), 22 transfer RNA genes (tRNAs), 13 protein-coding genes (PCGs) and two typical non-coding control regions (control region (CR) and origin of the light strand (OL)) with regulatory elements essential for transcription and replication1, 2. Because of the characteristics of coding content conservation, maternal inheritance, rapid evolution, and low levels of intermolecular genetic recombination, mitogenomes have become increasingly effective and popular markers for molecular research, such as phylogenetic molecular evolution, population genetics, phylogenetics, and comparative and evolutionary genomics3, 4. In addition to comparing nucleotide and amino acid sequence applying to molecular evolution, the complete mitogenome of tRNA secondary structure, gene rearrangement and models of the control of replication and transcription have been used extensively for deep-level phylogenetic inference in taxonomy in recent decades5, 6.
Scorpaeniformes, whose members are known for their commercial importance (e.g., Sebastes, Ophiodon) and venomous spines (e.g., Pterois, Scorpaena), is one of the largest and most morphologically diverse teleostean orders of fish, with more than 1400 species classified in 38 families depending on the taxonomy7, 8. Due to commercial overfishing9, four scorpionfishes have been protected by the International Union for Conservation of Nature (IUCN). In addition, some of Scorpaeniform fishes, such as Synanceiidae (stonefishes), have caused numerous human injuries due to their venom10,11,12. Several partial mitochondrial gene sequences of the 12S rRNA, the 16S rRNA and the complete tRNA-Val and nuclear genes of the large ribosomal subunit (28S), histone H3, and TMO-4c4 from Scorpaeniform have been sequenced and extensively used for phylogenetic analysis13. Nevertheless, the short genes do not provide enough information for phylogenetic relationships and sometimes result in controversial signals14, 15. Triglidae fishes belong to a moderately large family of the order Scorpaeniform, comprising approximately 14 genera and 100 species in tropical and temperate waters of the world’s oceans16. Because of medical importance and commercial value, many researchers have studied some aspects of Triglidae’s ecological distribution, morphology and biological habits17,18,19, but few investigations have focused on the taxonomy based on molecular features.
To date, more than 100 complete mitogenomes from teleostean species have been sequenced, but only 28 species in 5 families from Scorpaeniform are available in GenBank, which does not include the family Triglidae. The gurnards Chelidonichthys kumu and Lepidotrigla microptera are two species of the family Triglidae and are distributed in New Zealand, Australia, Japan, Malaysia, South Korea and China. However, the complete mitogenomes of C. kumu and L. microptera have not been determined. To understand the higher-level relationships of Scorpaeniformes, in this study, we sequenced the complete mitochondrial genome of the two Triglidae species and investigated the gene content and organization compared with other species. Furthermore, we reconstructed a phylogenetic tree based on PCG sequences for the purpose of analyzing the evolutionary relationships among Scorpaeniformes fish. In addition, the characterization of the C. kumu and L. microptera mitogenomes may provide more insight into the genesis and mitogenome evolution of teleostean species.
Methods
Sampling and DNA extraction
The specimens of C. kumu and L. microptera, which did not involve endangered or protected species according to the IUCN Red List, were collected in the Pearl River estuary (N 21°57′, E 133°47′), China, in July 2016. Our study was conducted with the approval from the Institutional Animal Care and Use Committee at Jinan University. All operations were performed according to international guidelines concerning the care and treatment of experimental animals. All samples were preserved in 95% ethanol and were stored at −80 °C until DNA extraction. Total genomic DNA was extracted from dorsal muscle tissue samples using the Animal Tissue Genomic DNA Extraction Kit (SangonBiotech, China). Extracted DNA was used to amplify the complete mitogenomes by PCR.
PCR amplification and sequencing
For the amplification of the C. kumu and L. microptera mitogenomes, several primer pairs were designed based on the aligned mitogenome sequences of Scalicus amiscus (GenBank: AP004441.1) (Table S1)20. The PCR amplifications were performed with LA Taq DNA polymerase using Premix LA Taq (Takara, China) under the following conditions: an initial denaturation step at 95 °C for 3 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 50 °C for 30 s and elongation at 72 °C for 1–5 min. All the PCR products were sent to Beijing Genomics Institute and sequenced using the primer walking method with a 3730XL DNA Analyzer. The obtained sequences had 100% coverage of the PCR products.
Sequence analysis
The sequences were manually checked and assembled with the program Seqman within Lasergene software. The final complete sequence annotation was performed using NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast) and the DNAStar package (DNAStar Inc. Madison, WI, USA). The location of the two rRNAs and the 13 PCGs for each species were primarily identified through Dual Organellar Genome Annotator (DOGMA)21. The majority of the transfer RNA (tRNA) genes were identified by the tRNA-scan-SE1.21 from the website http://lowelab.ucsc.edu/tRNAscan-SE/ using the default search mode and the ‘Mito/chloroplast’ source22. To infer the secondary structures of tRNA molecules, we used a commonly accepted comparative approach to correct for unusual pairings with RNA-editing mechanisms that are well known in fish mitogenomes. The software RNAstructure was used to draw the secondary structure of tRNA genes and the origin of light strand replication (OL)23. The codon usage of the 13 PCGs was calculated using MEGA 624. Tandem repeats in the control region (CR) were analyzed using the Tandem Repeats Finder program (http://tandem.bu.edu/trf/trf.html)25. The nucleotide composition skewness was measured according to the following formulas: AT skew [(A − T)/(A + T)] and GC skew [(G − C)/(G + C)]26.
The complete mitochondrial genomic DNA sequence of the C. kumu and the L. microptera were deposited into the GenBank database under the accession numbers KY379222 and KY012348, respectively.
Phylogenetic analysis
To investigate the phylogenetic relationships among fish, a total of 27 Scorpaeniformes mitogenomes available in GenBank were used (Table 1). The mitogenomes of Perciformes fish (Caesio cuning, Emmelichthys struhsakeri and Banjos banjos) were used as outgroups. The amino acid sequences and nucleotide sequence for each species of the 13 PCGs were aligned using default settings and concatenated, which were used for phylogenetic analysis via the Maximum Likelihood (ML) and Bayesian inference (BI) methods using raxmlGUI and MrBayes v 3.2.4, respectively27, 28. Every gene was aligned separately using Clustal X with default settings29. GTR+ I+ G was selected as the appropriate model for the nucleotide sequences by Modeltest 3.7 based on Akaike’s information criterion (AIC)30. MtArt+ I+ G+ F was the appropriate model for the amino acid sequence dataset according to ProtTest 3.4 based on AIC31. Four independent runs were allowed to run simultaneously for 1,000,000 generations and each was sampled every 1,000 generations, with the first 25% discarded as burn-in. Stationarity was considered to be reached when the average standard deviation of split frequencies was much less than 0.01. The resulting phylogenetic trees were drawn in FigTree v1.4.3.
Results and Discussion
Genome organization and structure
The complete mitochondrial genome sequences of C. kumu and L. microptera are 16,495 bp and 16,610 bp in length. Both of them are closed circular molecules. The two mitogenomes are typical of other Scorpaeniformes fish mitogenomes, including 13 PCGs (cox1–3, nad1–6, nad4L, atp6, atp8 and cob), 2 rRNA genes (12S rRNA and 16S rRNA), 22 transfer RNA genes (one for each amino acid and two each for serine and leucine) and two non-coding regions (OL and the control region (CR)) (Fig. 1). Among these 37 genes, twenty-three genes are transcribed on the heavy strand (H-strand), except for ND6 and eight tRNA genes (Gln, Ala, Asn, Cys, Tyr, Ser (UCN), Glu, and Pro). The order and orientation of genes in the C. kumu and L. microptera mitogenomes are identical to the other Scorpaeniformes species sequenced to date20, 32, 33.

Circular map of the mitogenome of C. kumu (A) and L. microptera (B). Transfer RNAs are designated by the IUPAC-IUB single letter amino acid codes (L1: trnLCUN; L2: trnLUUR; S1: trnLAGN; S2: trnLUCN) Genome organization names not underlined indicate coding sequence on the heavy strand and those with underline indicate coding sequence on the light strand.
Skewness, overlapping, and intergenic spacer regions
The mitogenome of C. kumu has a 48 bp overlap between genes in ten locations ranging from 1 to 20 bp, with the longest 20 bp overlap located between 16S RNA and trnL. In seven locations of the L. microptera mitogenome, genes overlapped by 1 to 50 bp, and the longest 50 bp overlap is located between ATPase 8 and ATPase 6. The mitogenomes of C. kumu contains 65 bp of intergenic spacer sequences (1 to 36 bp in length) spread over 11 regions, while there are 78 bp nucleotides dispersed in 10 intergenic spacers, ranging in size from 1 to 38 bp, in the mitogenomes of L. microptera. The longest spacer sequences in the two species are located between the trnN and the trnC, which are extremely C + G rich. The nucleotide compositions of the H-strand of C. kumu and L. microptera mitogenomes are as follows: A = 4,393(26.6%), T = 4,156 (25.2%), G = 2,811 (17.0%), and C = 5,135 (31.1%); A = 4,407(26.5%), T = 4,161 (25.1%), G = 2,857 (17.2%), and C = 5,185 (31.2%), respectively. The total nucleotide composition of the C. kumu and L. microptera mitogenomes are 51.8% and 51.6% A + T rich, respectively. Compared with other Scorpaeniformes species, the enrichments are lower than many species sequenced, i.e. I. spatula (52.5%), H. lagocephalus (53.2%), C. szanaga (52.7%), C. dzungaricus (53.2%), S. amiscus (54.4%), S. marmoratus (55.3%) and S. verrucosa (59.3%). In contrast, these enrichments are slightly higher compared with those of C. poecilopus (51.4%) and Cottus hangiongensis (51.3%). Both of the highest A + T contents (60.9% and 59.8%) were detected in the control region, which is consistent with previous research on other fish20, 34. Additionally, the AT skew (0.0277) for the C. kumu mitogenome is slightly positive, indicating a higher occurrence of As to Ts, and its GC skew (−0.2925) is negative, indicating a higher content of Cs than Gs. The AT skew (0.0287) and GC skew (−0.2895) for the L. microptera mitogenome are identical to those of the L. microptera mitogenome. These findings are similar to all sequenced Scorpaeniformes mitogenomes to date, except for Cottus poecilopus, Cottus hangiongensis and Satyrichthys amiscus 20, 34, which have a negative AT skew.
Protein-coding genes
The total nucleotide length of the 13 PCGs in C. kumu and L. microptera are 11,429 bp and 10,450 bp, respectively. The start and stop codons of the 13 PCGs in the C. kumu and L. microptera mitogenomes are shown in Table 2. Methionine (ATG) is the start codon for most PCGs genes, except for cox1, which utilizes GTG, an accepted canonical mitochondrial start codon for vertebrate mitogenomes35,36,37. The stop codons (TAA, TTA and TAG) and one incomplete stop codon (T) are utilized in these PCGs. In C. kumu, ND2 ends with TTA, ND3 ends with TAG, three genes (cox2 ND4 and cytb) use T and the remaining genes terminate with TAA. The stop codons of the two fish mitogenomes are identical, with the exception of T and TAG as stop codons for ATP8 and ND5, respectively, in L. microptera. The incomplete stop codon is usually found in metazoan mitogenomes, which is presumably completed via post-transcriptional polyadenylation38. Relative synonymous codon usage values for the C. kumu and L. microptera mitogenomes are summarized in Table S2 and Fig. 2. The total number of codons in PCGs of C. kumu is 3808, where AGA and AGG codons are not represented. In the L. microptera mitogenome, the 13 PCGs have full codons (total number: 3431). Leucine 1 (Leu 1, 586), alanine (Ala, 351), and threonine (Thr, 303) are the most common amino acids in C. kumu mitogenome PCGs. In L. microptera mitochondrial proteins, the three amino acids (Leu 1, 535; Ala, 297 and Thr, 294) are the most common. The A + T content of the 13 PCGs of C. kumu and L. microptera mitogenomes is 50.6% and 50.5%, respectively. Furthermore, the AT skew and GC skew values of the PCGs of the two species are shown in Fig. 3. The most AT skew is negative, except for five genes (cox2, ATP8 for C. kumu and ND2, cox2, ATP8 and ND6 for L. microptera). All GC skew values are negative with the exception of the ND6 gene of C. kumu, which had a positive GC skew. This result suggests that more Ts and Cs are present in most PCGs, which is consistent with most previous observations34, 39.

The relative synonymous codon usage (RSCU) in the mitogenomes of C. kumu and L. microptera.

Graphical illustration showing the AT and GC skew in the PCGs of the mitochondrial genome of C. kumu (A) and L. microptera (B).
Transfer RNA genes and ribosomal RNA genes
The predict result of 22 tRNA genes (two for Serine and Leucine and one for each of the other amino acids) in the two fish mitogenomes are showed from Fig. 4. Fourteen tRNAs are encoded by the H-strand, and the remaining 8 tRNAs are encoded by the L-strand (Table 2). All tRNAs varied in size from 62 bp (trnC) to 74 bp (trnL1 (UUR) and trnK) in C. kumu and ranged from 65 bp (trnC) to 74 bp (trnL1 (UUR) and trnK) in L. microptera. This tRNA genomic architecture is identical to all Scorpaeniformes species examined to date39, 40. All of the 22 tRNA genes could be folded into typical cloverleaf secondary structures with the exception of trnS2 (AUN). The secondary structure of trnS2 (AUN) lacks a DHU stem, and this phenomenon has been observed in many animals of metazoan mitogenomes including Scorpaeniformes species39, 40. A 7 bp amino acid acceptor stem is conserved in all tRNAs, except for trnaV and trnaL1 (UUR) in L. microptera, which has a 9 bp amino acid acceptor stem. Additionally, 28 to 33 unmatched base pairs are found in the secondary structure of all tRNAs in both fish mitogenomes. All of the unmatched base pairs are G-U pairs, which formed a weak bond, except for the U-U pair in the TΨC stem of Leu2 in C. kumu. A positive AT skew (0.1127 and 0.1193) and a negative GC skew (−0.1011 and −0.1067) are found among the concatenated sequence of all 22 tRNAs in C. kumu and L. microptera respectively, indicating A nucleotides are more likely to occur than Ts, so are Cs nucleotides than Gs. A similar result occurred in the ribosomal genes in both species. In mitogenomes of C. kumu, the 12S and 16S rRNA genes are 946 bp (51.0% A + T content) and 1653 bp (53.1% A + T content), respectively. As in mitogenomes of L. microptera, the length of 12S rRNA is 1041 bp with an A + T content of 51.2% and the 16S RNA is 1695 bp with an A + T content of 53.2%. The two rRNAs are located between trnF and trnL1 (UUR). They are separated by the trnV. The location was the case in most vertebrates34, 35, 39, 40.

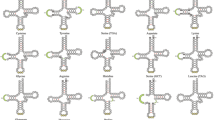
Predicted secondary structures for the tRNA genes in the C. kumu and L. microptera mitogenome.
Non-coding regions
Except for the several short non-coding regions between genes, there are also two long non-coding regions in the mitochondrial genomes of C. kumu and L. microptera: OL and CR, which are important in the transcription, replication and maintenance of the mitochondrial genome41. In mitogenomes of C. kumu and L. microptera, OL is found in the domain between trnN and trnC, which is folded a hairpin secondary structure (Fig. 5). The length of C. kumu is 36 bp in C. kumu and 38 bp in L. microptera, respectively. The control region is essential for the initiation of replication in vertebrates, which is found between tRNAPro and tRNAPhe. The CR in C. kumu spanned 838 bp with a 60.8% A + T content and shows an equal AT skew (0) and a negative GC skew (−0.0793). The CR in L. microptera spanned 838 bp with 59.3% A + T content and shows a slightly positive AT skew (0.0181) and a negative GC skew (−0.1144). The A + T content of the two mitogenomes is lower than most of the other Scorpaeniformes species to date33, 34. Several conserved sequence blocks (CSBs) are observed in the control regions of teleost fish, which could play important roles in mitochondrial metabolism42. In CR of teleost fish, CSB-D, -E and -F are found in central conserved domains. Meanwhile, CSB-1, CSB-2 and CSB-3 are typically present in the conserved sequence block domain42. All the CSBs are identified in C. kumu and L. microptera, by comparing them with the recognition sites in Scorpaeniformes species (Fig. 6). Both species have no relatively similar repetitive motifs in the control regions, which indicated that the repeat motifs occurred after family diversification. However, more information is necessary to further evaluate evolutionary aspects of repeat clusters. In addition, tandem repeats are not found in the two species using the Tandem Repeats Finder program25. This phenomenon exists for other Scorpaeniformes species with the exception of the genus Sebastes 33, 34.

The putative hairpin secondary structure of the OL found in the C. kumu (A) and L. microptera (B) mitogenome.

Features present in the control regions of the C. kumu (A) and L. microptera (B) mitogenomes. The conserved sequence blocks contain CSB-F (red), CSB-E (purple), CSB-D (orange), CSB-1 (deep blue), CSB-2 (green) and CSB-3 (light blue). The conserved motifs ATGTA and its complement TACAT which may form thermostable hairpin structure are shaded.
Phylogenetic analysis
The interrelationship of Scorpaeniformes remains contentious due to the lack of corroborative evidence43. In this study, six Scorpaeniformes families of the 13 PCGs from mitogenomes, concatenated nucleotide sequences and amino acids, are used to reconstruct phylogenetic relationships via the BI and ML methods (Figs 7 and S1). The results provide good support for the monophyly of each family. The phylogenetic trees include 29 Scorpaeniformes species, representing 7 families and 11 genera. The best supported phylogenetic relationship found in this study is as follows: (Synanceiidae + (Scorpaenidae + Sebastidae)) + ((Peristediidae + Triglidae) + (Cottidae + Hexagrammidae)). We sequenced two species (C. kumu and L. microptera) within Triglidae, which formed a monophyletic group with Peristediidae. The analysis suggests that Peristediidae are most closely related to Triglidae. The topology of the relationships of Scorpaeniformes is similar to previous work13, and more mitogenomes from Triglidae fish are needed for further phylogenetic analyses and to demonstrate the relationships among these families in the future.

Phylogenetic trees inferred from amino acid and nucleotide sequences of 13 PCGs of the mitogenome using BI analysis. Perciformes fish (C. cuning, E. struhsakeri and B. banjos) were used as outgroups. The numbers along branches indicate posterior probability values (red for amino acid and blue for nucleotide sequences). The accession numbers are shown behind the species names.
Conclusion
The first two complete mitochondrial genomes (C. kumu and L. microptera) for the family Triglidae were determined and compared with those of other Scorpaeniformes species. The mitogenome sequences of C. kumu and L. microptera were 16,495 bp and 16,610 bp in length, respectively. Each mitogenome is composed by the typical structure of 13 PCGs, 2 rRNAs, 22 transfer RNA genes and two non-coding regions. Both of the complete mitogenomes of Triglidae fish were typical circular molecules and had similar genome organization and structure as those found in other teleostean species. Similar to other vertebrate mitogenomes, most of the PCGs utilized ATG, except for cox1. Additionally, all of the tRNAs could be folded into typical cloverleaf secondary structures with the exception of trnS2 (AUN), which lacks a DHU stem. Several CSBs were found in the control regions of the two fish, which could play important roles in mitochondrial metabolism. By phylogenetic analysis, the mitogenome sequences could be used to resolve the higher-level relationship of Scorpaeniformes. The results showed that Triglidae are most closely related to Peristediidae. Our findings provided important data for further studies on the population genetics and evolutionary biology of Scorpaeniformes. Additional taxonomic work is needed in the future.
References
Miya, M. & Nishida, M. Organization of the mitochondrial genome of a deep-sea fish, gonostoma gracile (Teleostei: Stomiiformes): First example of transfer RNA gene rearrangements in bony fishes. Mar Biotechnol 1, 416–0426 (1999).
Shi, X., Tian, P., Lin, R., Huang, D. & Wang, J. Characterization of the complete mitochondrial genome sequence of the globose head whiptail Cetonurus globiceps (Gadiformes: Macrouridae) and its phylogenetic analysis. Plos One 11, e0153666 (2016).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Research 27, 1767–1780 (1999).
Bentzen, P., Wright, J. M., Bryden, L. T., Sargent, M. & Zwanenburg, K. C. Tandem repeat polymorphism and heteroplasmy in the mitochondrial control region of redfishes (Sebastes: Scorpaenidae). J Hered 89, 1–7 (1998).
Wang, Z. L., Li, C., Fang, W. Y. & Yu, X. P. The complete mitochondrial genome of two retragnatha spiders (Araneae: Tetragnathidae): Severe truncation of tRNAs and novel gene rearrangements in araneae. Int J Biol Sci 12, 109–119 (2016).
Liu, Q. N. et al. The first complete mitochondrial genome for the subfamily Limacodidae and implications for the higher phylogeny of Lepidoptera. Sci. Rep 6, 35878 (2016).
Nelson, J. S. Fishes of the world. 3rd ed. Wiley, New York (1994).
Washington, B. B., Eschmeyer, W. N., Howe, K. M. Scorpaeniformes: relationships In Ontogeny and Systematics of Fishes, American Society of Ichthyologists and Herpetologists Special Publication (Eds Moser, H. G., Richards, W. J. et al.) 438–447 (Lawrence, 1984).
Markle, D. F. The Rockfishes of the Northeast Pacific. Copeia 2, 421–422 (2001).
Burnett, J. W. Aquatic adversaries: stonefish. Cutis 62, 269–270 (1998).
Halstead, B. W., Vinci, J. M. Biology of poisonous and venomous marine animals In Marine toxins and venoms and Handbook of Natural Toxin (Ed. Tu, A. T.) 1–30 (1988).
Kizer, K. W., Mckinney, H. E. & Auerbach, P. S. Scorpaenidae envenomation. A five-year poison center experience. Jama-J Am Med Assoc 253, 807–810 (1985).
Smith, W. L. & Wheeler, W. C. Polyphyly of the mail-cheeked fishes (Teleostei: Scorpaeniformes): evidence from mitochondrial and nuclear sequence data. Mol Phylogenet Evol 32, 627–646 (2004).
Fernández, R., Hormiga, G. & Giribet, G. Phylogenomic analysis of spiders reveals nonmonophyly of orb weavers. Curr Biol 24, 1772–1777 (2014).
Wheeler, W. & Hayashi, C. The phylogeny of extant chelicerate orders. Cladistics 14, 173–192 (1998).
Richards, W. J. & Jones, D. L. Preliminary classification of the gurnards (Triglidae: Scorpaeniformes). Mar Freshwater Res 53, 274–282 (2002).
Richards, W. J. & Saksena, V. P. Systematics of the Gurnards, Genus Lepidotrigla (Pisces, Triglidae), from the Indian Ocean. B Mar Sci 27, 208–222 (1977).
Colloca, F., Cardinale, M. & Ardizzone, G. D. Biology, spatial distribution and population dynamics of Lepidotrigla cavillone (Pisces: Triglidae) in the Central Tyrrhenian Sea. Fisheries Research 32, 21–32 (1997).
Terrats, A., Petrakis, G. & Papaconstantinou, C. Feeding habits of Aspitrigla cuculus (L., 1758) (red guirnard), Lepidotrigla cavillone (Lac., 1802) (large scale gurnard) and Trigloporus lastoviza (Brunn., 1768) (rock gurnard) around Cyclades and Dodecanese Islands. Mediterr Mar Sci 1, 91–104 (2000).
Miya, M. et al. Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences. Mol Phylogenet Evol 26, 121–138 (2003).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Mathews, D. H. Using the RNAstructure Software Package to Predict Conserved RNA Structures. Curr Protoc Bioinformatics 46, 12.4.1–12.4.22 (2014).
Lewis, P. O., Kumar, S., Tamura, K., Nei, M. & Lewis, P. O. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Bioinformatics 30, 2725–2729 (1994).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol 41, 353–358 (1995).
Silvestro, D. & Michalak, I. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12, 1–3 (2012).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61, 539–542 (2012).
Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Current Protocols in Bioinformatics Chapter 2, 2.3.1–2.3.22 (2002).
Nylander, J. MrModeltest v2. Program distributed by the author, vol. 2. Evolutionary Biology Centre, Uppsala University (2004).
Abascal, F., Zardoya, R. & Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 (2005).
Wu, Z., Wang, D., Hu, J. & Wang, Q. Complete mitochondrial genome of the Scorpaenopsis cirrhosa (Scorpaeniformes: Scorpaenidae). Mitochondr DNA 27, 3449–3450 (2016).
Wang, Q., Wang, J., Luo, J. & Chen, G. The complete mitochondrial genome of the Synanceia verrucosa (Scorpaeniformes: Synanceiidae). Mitochondr Dna 6, 4234–4235 (2015).
Hwang, D. S., Byeon, H. K. & Lee, J. S. Complete mitochondrial genome of the freshwater sculpin Cottus koreanus (Scorpaeniformes, Cottidae). Mitochondr DNA 24, 674–675 (2013).
Xiao, X. et al. The complete mitochondrial genome and phylogenetic analysis of Chinese Jianchang horse (Equus caballus). Clon Transgen 5, 149 (2016).
Yue, G. H., Liew, W. C. & Orban, L. The complete mitochondrial genome of a basal teleost, the Asian arowana (Scleropages formosus, Osteoglossidae). BMC Genomics 7, 242 (2006).
Yang, X. et al. Characterization of the complete mitochondrial genome sequence of Homalogaster paloniae (Gastrodiscidae, Trematoda) and comparative analyses with selected digeneans. Parasitol Res, 1–9 (2016).
Ojala, D., Montoya, J. & Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 (1981).
Jang, Y. S. et al. The complete mitochondrial genome of the dark-banded rockfish Sebastes inermis (Scorpaenidae, Scorpaeniformes). Mitochondr DNA 26, 895–896 (2015).
Li, Y., Shi, Y., Lu, J., Ji, W. & Wang, Z. Sequence and phylogenetic analysis of the complete mitochondrial genome of Lasiopodomys mandarinus mandarinus (Arvicolinae, Rodentia). Gene 593, 302–307 (2016).
Fernández-Silva, P., Enriquez, J. A. & Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol 88, 41–56 (2003).
Lee, W. J., Conroy, J., Howell, W. H. & Kocher, T. D. Structure and evolution of teleost mitochondrial control regions. J Mol Evol 41, 54–66 (1995).
Imamura, H. & Yabe, M. Demise of the Scorpaeniformes (Actinopterygii: Percomorpha):An Alternative Phylogenetic Hypothesis. Bulletin of Fisheries Sciences Hokkaido University 53, 107–128 (2002).
Acknowledgements
The authors would like to thank Dr. Xiaojing Wang and Dr. Chunmei Zhong of South China Normal University, Dr. Yadong Cui of Fuyang Normal University, and Tao Shi of Dalian Jiaotong University for their suggestions on the experiment operation. This work was supported by the National Natural Science Foundation of China (31372535 and 41576162), and the Fundamental Research Funds for the Central Universities of China.
Author information
Authors and Affiliations
Contributions
L.C. and S.-H.L. conceived and designed the experiments. X.-C.G. and J.-Y.C. contributed reagents and materials. L.C., H.Z., L.L. and F.-H.L. performed the experiments. L.C. and Y.-L.D. analyzed the data. Y.-L.D. prepared all figures. L.C. and Y.-L.D. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cui, L., Dong, Y., Liu, F. et al. The first two complete mitochondrial genomes for the family Triglidae and implications for the higher phylogeny of Scorpaeniformes. Sci Rep 7, 1553 (2017). https://doi.org/10.1038/s41598-017-01654-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01654-y
This article is cited by
-
Diagnostic applicability of mitogenomics in uncovering intraspecific carangid diversifications: insights into phylogeny, divergence time, and characterization of two cryptic Selaroides leptolepis mitogenomes
Organisms Diversity & Evolution (2024)
-
Characterization and comparison of the complete mitochondrial genomes of two stingrays, Dasyatis pastinaca and Dasyatis tortonesei (Myliobatiformes: Dasyatidae) from the Mediterranean Sea
Molecular Biology Reports (2021)
-
Characterization of the Complete Mitochondrial Genome of Arius dispar (Siluriformes: Ariidae) and Phylogenetic Analysis Among Sea Catfishes
Journal of Ocean University of China (2020)
-
Characterization of the Whole Mitogenome of Largehead Hairtail Trichiurus lepturus (Trichiuridae): Insights into Special Characteristics
Biochemical Genetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
