
Abstract
Amides are ubiquitous and abundant in nature and our society, but are very stable and reluctant to salt-free, catalytic chemical transformations. Through the activation of a “sterically confined bipyridine–ruthenium (Ru) framework (molecularly well-designed site to confine adsorbed H2 in)” of a precatalyst, catalytic hydrogenation of formamides through polyamide is achieved under a wide range of reaction conditions. Both C=O bond and C–N bond cleavage of a lactam became also possible using a single precatalyst. That is, catalyst diversity is induced by activation and stepwise multiple hydrogenation of a single precatalyst when the conditions are varied. The versatile catalysts have different structures and different resting states for multifaceted amide hydrogenation, but the common structure produced upon reaction with H2, which catalyzes hydrogenation, seems to be “H–Ru–N–H.”
Similar content being viewed by others
Introduction
As a result of high thermodynamic stability and kinetic inertness1,2,3, amides have been found in natural systems for millennia, as the repeating units of functional polypeptides (proteins), and have more recently become a valuable commodity as the monomer units (e.g., α, β-unsaturated carboxamides, caprolactams) of synthetic polymers including poly(acrylamide), nylons, and Kevlar produced on an enormous scale. Rapidly emerging C(sp3)–H and C(sp2)–H bond activation strategies that lead to C–N4, 5 and C–C6, 7 bond formation frequently utilize amides as directing groups. Were it possible to develop an effective method for the catalytic hydrogenation of amides (formally, a hydrogenolysis–hydrogen addition sequence) that leads to selective C–N bond cleavage in preference to C=O bond cleavage, alcohols and amines would be generated. Both are useful platform chemicals or intermediate building blocks for organic synthesis. Alcohol and amine monomer units could also be regenerated/recycled from waste polyamides, which would otherwise be disposed of via combustion, resulting in the emission of CO2 and NO x . For example, the hydrogenation leading to effective C–N bond cleavage in N, N-dimethyl formamide (DMF) produces CH3OH. This method, if it can be developed, would be potentially useful for enhancing the “anthropogenic chemical carbon cycle (methanol economy),” as suggested by Olah8, in conjunction with the elegant DMF synthesis reported by Noyori9 involving the reaction of supercritical CO2 with H2 and Me2NH, catalyzed by a ruthenium (Ru) complex with an extremely high turnover number (TON) (substrate/catalyst ratio: S/C = 370,000). A tandem stoichiometric amine- [(CH3)2NH-10 or 2-aminoethanol-11] and catalytic ruthenium (Ru)-promoted hydrogenation of CO2 to CH3OH was also reported.
Amide hydrogenation systems involving molecular catalysts developed separately thus far show different selectivity and reactivity, and are complementary to each other. Cole-Hamilton12, 13, Leitner/Klankermayer13,14,15, and Beller16 have developed different (triphos)Ru catalyst systems (triphos = CH3C[CH2PPh2]3; Ph = C6H5) for selective cleavage of the C=O bond of amides. When combined with catalytic Yb(OSO2CF3)3, hydrogen pressure (P H2) can be reduced as low as ca. 0.5 MPa16. Use of a combination of (P, C, P)Ir pincer complex and stoichiometric B(C6H5)3 was also proved to be effective for inducing the C=O bond cleavage17. In contrast, Ru complexes for the selective cleavage of the C–N bond of amides were intensively studied by Ikariya ((P, NH)Ru)18,19,20,21, Milstein ([P,(N, N)bpy]Ru, where bpy = bipyridine; (N, N)bpy = bipyridine nitrogens)22,23,24,25, Bergens ([(P, NH2)(P, NH2)]Ru)26,27,28, Leitner/Klankermayer, ((triphos)Ru)15, and Beller ((P,NH, N)Ru)29. Milstein’s milestone discovery, in which deprotonation at the 2-(pyridyl)methylene unit in the (P, N py, P)Ru pincer complexes induces an active catalyst30, has had a great impact on the ensuing molecular design of hydrogenation catalysts (py = pyridine; N py = pyridine nitrogen). Iron (Fe) complexes catalyze the C–N bond cleavage31, 32 with the aid of the tridentate (P, NH, P) ligand originally developed by Takasago Co. for ester hydrogenation33, 34. A (P, N py, P)Fe pincer complex showed comparable catalytic activity35. Unfortunately, however, those metal complexes hydrogenate majorly a range of strongly or moderately activated amides, including N-aryl-, N-acyl-, N-(di)methyl-, and α-alkoxy amides and morpholino ketones, as well as relatively simple and small amides (e.g., formamides, acetamides, trifluoroacetamides). Very recently, Zhang36 reported an extremely active (P, N py, NH, P)Ru catalyst that hydrogenates, in most cases, activated amides, where a rather low hydrogenation temperature (100 °C) was only tested. Mashima37 reported the use of a combination of a Ru complex (2 mol %) bearing two bidentate (P, NH2) ligands, KOtBu (20 mol %) and Zn salts (4 mol %), which promoted the hydrogenation of N-methyl amides at 100 °C, but the reactivity of sterically more demanding amides and primary amides was scant to moderate; the catalysis does not seem to be sustainable at elevated temperatures.
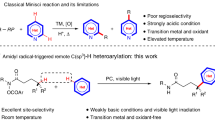
Thus, a possibility of a new robust and versatile catalyst system for amide hydrogenation that tolerates not only mild but also harsh (high reaction temperature (T) and high hydrogen pressure (P H2)) reaction conditions has remained elusive. An exploration of hydrogenation precatalyst, generally and potentially be applicable for both C–N and C=O bond cleavage, as well as for different classes of amides such as those found in DMF, oligopeptides and artificial polyamides, remains a grand challenge; however, there is a lack of basic knowledge concerning the molecular design of a structurally robust, multifunctional catalysts derived diversely from a single precatalyst competent under various reaction conditions necessary to achieve such multifaceted amide hydrogenation.
We recently reported preliminary research on the molecular design of [(P, N py) (P, N py)]Ru complexes including RUPCY (2a, Cy = C6H11)38,39,40,41, which was shown to be effective for the hydrogenation of unactivated amides38, 40, 41. Unfortunately, however, the catalyst system is only viable under harsh reaction conditions (S/C = 50; P H2 = 4–8 MPa; T = 140–180 °C, reaction time (t) = 24–216 h). Herein is reported a greatly improved method for the hydrogenation of unactivated amides (Fig. 1), realized by (P,(N, N)bpy, P)Ru complexes RUPIP2 (1a, iPr = (CH3)2CH) and RUPCY2 (1b) as precatalysts, from which versatile catalysts are generated by incorporation of multiple hydrogen atoms to a sterically confined bpy–Ru framework (molecularly well-designed site to confine adsorbed H2 in). Through simple but rational structural modification of 2a to 1a 41 and 1b 41, catalyst performance for C–N bond and C=O bond cleavage has been significantly advanced (TON up to 7700) under both mild (P H2 = 0.5–2 MPa; T = 60–120 °C) and harsh (P H2 = 3–8 MPa; T = 130–160 °C) conditions.

Summary of this work. Our earlier work (in the upper square) illuminated possible catalysts I A and I B derived from 2a, which is modified to new Ru complexes 1a and 1b (in the lower square); and the X-ray single crystal structure of 1b. Ru (black), Cl (green), N (blue), P (red), C (grey), H (white).
Results and Discussion
In our previous study38, it was shown that a plausible catalyst, either I A or I B , was likely induced upon treatment of precatalyst 2a with bulky base 2b and H2 (Fig. 1). Under the preactivation conditions for 2a, full hydrogenation of the pyridines of 2a gave a new bidentate ligand 2c incorporating the piperidine (N–H) unit. It is therefore likely that hydrogen(s) transfer takes place from the four-centered “H–Ru–N–H” unit to an amide carbonyl group (Noyori’s bifunctional mechanism)42,43,44, affording a N, O-hemiacetal R1CH(OH)(NR2R3). Thus, this amide hydrogenation mechanism may involve an outer-sphere, rather than inner-sphere, mechanism that involves a two-step pathway, wherein the Ru catalyst having an H–Ru–N–H functionality is generated in the first step, followed by the amide carbonyl group interacting with both of the H of the H–Ru–N–H component to facilitate the hydrogen (H− and H+) transfer. However, the question as to which catalyst (I A or I B ) is more active for hydrogenation has yet to be answered. To probe this, Ru complex 1b bearing a tetradentate bpy analog ligand was synthesized, in which the two bidentate ligands of 2a are connected. This simple ligand manipulation rules out the possibility of facile detachment of the ligand from the Ru center, which the bidentate ligand 2c of I A and I B underwent. According to the X-ray single crystal structure analysis of 1b (Fig. 1, ball-and-stick models; additional structural parameters are available in Supplementary Fig. 11), there are fifteen (non-H) atoms (the bpy, Ru, and two phosphorus atoms) in a planar orientation constituting a “sterically confined bpy–Ru framework” with four sterically bulky cyclohexyl (Cy) architectures on top and bottom. The sterically encumbered H2-adsorption site could preferentially confine the small molecule in, rather than more sterically bulky substrates such as amides. Steric repulsion between an incoming substrate and the four Cy groups reasonably suppresses substrate–Ru interactions that may cause catalyst inhibition and/or decomposition of catalysts.
At the outset, the performance of 1b as catalyst was compared with that of 2a (Fig. 2a): toluene solutions of each Ru complex (1 mol %), amide 3a (100 mol %), and NaH (6 mol %) were reacted under identical reaction conditions ([Ru]0 = 3.3 mM, S/C = 100; P H2 = 1 MPa, T = 110 °C, t = 15 h). The apparent reaction rate obtained using 1b (4a: 82%) was more than 40-fold faster than that with 2a (4a: <2%) under mild conditions (entry 1 vs. 3). By replacing 1b with 1a, the initial load of the Ru complex can be reduced to not less than 0.25 mol % (S/C = 400; NaH: 5 mol %), giving 4a and 5a in 73% and 74% yield, respectively (Supplementary Table 2, entry 2; a TON of ca. 300, calculated as product (mol)/1a (mol)) ([1a]0 = 0.83 mM, P H2 = 1 MPa, T = 110 °C, t = 24 h). The catalytic activity of 1a and 1b exceed the best reported values obtained under similar conditions by a wide margin (4a: 57%: S/C = 100, P H2 = 1 MPa, T = 110 °C, t = 48 h, TON = 57)22. From this, the turnover frequency (TOF = TON · h−1) can be roughly estimated to be not more than 10-fold greater in this new system (300/24 vs. 57/48). In the meantime, remarkably high TON of 8800 was observed in the hydrogenation of 3a catalyzed by a (P, N py, NH, P)Ru complex, albeit with much higher P H2 (P H2 = ca. 5 MPa, t = 20 h)36.

Precatalysts tested and diverse catalysts derived from 1a. (a) Hydrogenation of 3a using different Ru-complexes 1a–f and 2a ([Ru]0 = 3.3 mM) as precatalysts. (b) Major resting states of catalysts. Calculated exact masses: I C1 –I C3 (519.1626), I C -d 2 (521.1752), I D (534.2661), I E (429.1603) and I E -d 16 (445.2608).
In order to find a more competent catalyst, several derivatives of 1 (1c 41, 45, 1d 41, 1e 41, and 1f 46) were synthesized as different precatalysts and tested in the hydrogenation of 3a under similar conditions ([1]0 = 3.3 mM, S/C = 100; 1:NaH:3a (mol %) = 1:6:100; P H2 = 1 MPa, T = 110 °C, t = 15 h) (Fig. 2a)41. The phenanthroline series 1d–f, which has a more extended π-conjugation system as H2-adsorbent, was totally ineffective (entries 5–7). The sterically more bulky 1c (R = tBu) also showed less promising results (entry 4). In order to minimize steric repulsion between the catalyst and the incoming substrate, including less reactive, sterically bulky amides, 1a was chosen for further investigation.
Bulky base 2b (Fig. 1), which was used previously38 as an additive for the preactivation of 2a in the absence of amide, was as effective as NaH for the amide hydrogenation catalyzed by 1a and 1b. Hydrogenation of three different, rather activated amides was also tested using 1a (0.01 or 0.1 mol %) preactivated with 2b or NaH (0.1 or 1 mol %) under H2 pressure (P H2(pre)) = 1 or 8 MPa and temperature (T pre) = 160 °C, and the results were compared with those reported by Bergens27, in which [Ru(Ph2P(CH2)2NH2)2(η3-allyl)]+(BF4 −) was used with a larger amount of a base (4 mol % of KN[Si(CH3)3]2 or 5 mol % of NaOCH3) (Supplementary Table 1 for reaction conditions and comparison of hydrogenation results). Higher to comparable TON and TOF under identical hydrogenation conditions (P H2 = 5 MPa, T = 100 °C, t = 24 h) were consistently shown by 1a in the hydrogenation of N, N-dimethylacetamide (TON = 530 vs. 500), N-phenylacetamide (TON = ~800 vs. 700), and N-phenyl-2-pyrrolidone (TON = 7700 vs. 7120) under more neutral pH. It turned out, however, that this preactivation step in the absence of amide could be eliminated using NaH. The resting state of a matured catalyst generated upon activation of 1a in the presence or in the absence of amide 3a ([1a]0 = 3.3 mM in toluene; 1a:NaH (mol %) = 1:10; [3a]0 = 5.0 mM or 0; P H2(pre) = 1 MPa, T pre = 110 °C, t pre = 5 h) was thus evaluated. Solutions of catalyst (1a activated with and without 3a) were prepared separately, and the samples were measured directly via electrospray ionization-mass spectroscopy (ESI-MS) (Supplementary Figs 1 and 2). Both samples showed a Ru species that retained a tetradentate ligand-incorporated structure during the induction period of the catalyst: organic frameworks comprised of a Ru with slightly different molecular weights (m/z = 519.1626 ± 0.0052 (strong intensity), 565.1317 ± 0.0021 (I C − 2 H + 3 O: small to moderate intensity)) were consistently detected. Both signals correspond to structures less the two Cl groups from the original 1a. The former intense signal corresponds to I C1 –I C3 (1H NMR (ppm): δ −8.62 (t (dd), J = 29.7 Hz, RuH)), produced by deprotonation of two methylene groups (CH2–bpy–CH2) of 1a (with two NaCl formation), followed by H2 adsorption (Fig. 2b). The deprotonation30, 38 and subsequent capture of one H2 molecule was confirmed using D2 instead of H2 (Supplementary Fig. 3). The 1H NMR signal at δ −8.62 became significantly weaker, and the ESI-MS signal of I C1 –I C3 disappeared; instead, a signal at 521.1752 ± 0.0070 (I C − H2 + D2) was observed as the only intense signal. In contrast, when activation of 1a was carried out under the identical conditions, except for P H2(pre) = 8 MPa and T pre = 160 °C or P H2(pre) = 4 MPa and T pre = 140 °C being used, the base peak (m/z = 429.1603 ± 0.0016) was consistent with I E (or its tautomers): fully hydrogenated bipyridine with concurrent cleavage of one C–P bond (Supplementary Figs 4–6)47, 48. The corresponding I E -d 16 (m/z = 445.2608 ± 0.0090) was also detected using D2 instead of H2, suggesting hydrogenolysis of the C–P bond. Under milder conditions (P D2(pre) = 1 MPa, T pre = 160 °C), I D (m/z = 534.2661 ± 0.0019 or its tautomers) was also detected as a major product (Supplementary Figs 7 and 8) which would correspond to an intermediate during the structural changes from I C to I E , during which time deuterium atoms from multiple D2 (one to eight molecules) are in turn incorporated. The oxygenation of I E likely occurred during aerobic sample injection into the ESI-MS instrument38, so that any oxygen atom-incorporating processes affecting the hydrogenation steps could be fully ruled out. These experiments show that different catalyst resting states, in which the common structure produced upon reaction with H2 is “H–Ru–N–H” (Fig. 2b), are generated to catalyze hydrogen transfer. The hydride- and proton-transfer to an amide carbonyl most likely occurs from a “H(δ−)–Ru–N–H(δ+)” fragment, in which both the Ru–H and N–H face the same direction. To summarize, boundary conditions as to whether multiple hydrogen addition would occur to the H2 adsorption site of 1a depend on both P H2(D2)(pre) and T pre. When a lower P H2(D2)(pre) = 1 MPa was used, intermediate I C was the major species with T pre = 110 °C, while I C was multiply in turn hydrogenated (T pre < 160 °C), giving I E (-d 16) as a minor species through the formation of partially hydrogenated I D . On the other hands, with P H2(pre) = 4–8 MPa and T pre = 140–160 °C, I C was converted more rapidly into I E as a major or exclusive species. The diverse mechanism hidden behind the activation process of 1a would be entirely different from that in the ester hydrogenation poorly promoted by 1f 46 and effectively catalyzed with its relevant Ru complexes bearing tetradentate (N,(N, N)bpy, P) ligands (P H2 = 5–10 MPa, T = 25–100 °C) reported by Zhou45, in which a possibility of multiple hydrogen additions to precatalyst was not totally investigated under milder T. The scant catalytic activity derived from phenanthroline series 1d–f could partly be explained by a more acidic nature of the NH group of aniline structures in the catalyst induced by activation of 1d–f (Supplementary Figs 9 and 10; Supplementary Table 2, entry 3). In any event, the H2-adsorption site with bulky iPr (or Cy) groups is non-innocent and undergoes meaningful structural changes via multiple hydrogen additions before producing the diverse, active forms of catalyst. These results further suggest that a Ru complex bearing a (P,(N, N)bpy, P) ligand is more versatile and robust in the production of diverse catalysts than Ru precatalysts bearing simple bidentate (P, N py)38, 40 and (P, NH)18,19,20,21, 26,27,28, 37, tridentate (P,(N, N)bpy)22,23,24,25, and tetradentate (P, N py, NH, P)36 ligand(s).
Hydrogenation can be started conveniently by mixing air-stable 1a, amide 3, and NaH together, followed by pressurizing the reaction vessel with H2 and then elevating T. Catalyst preactivation procedures in a separate reaction vessel is not necessarily needed. The hydrogenation of various unactivated amides under different conditions was tested, and the results are given in Figs 3 and 4 (Supplementary Table 2 gives detailed reaction conditions). Primary, secondary, and tertiary amides showed excellent compatibility with the same precatalyst, regardless of steric demands or whether aromatic/aliphatic. In order to shorten the reaction time for practical application, it is better to slightly increase the T to 120–130 °C, and P H2 to 2–3 MPa (Fig. 3a). The hydrogenation of ε-caprolactam (3h), a cyclic amide which serves as the monomer of nylon-6, showed a similar pattern of C–N bond cleavage, giving amino alcohol 6h predominantly (azepane: 1%). For the hydrogenation of unactivated amide 3h, the catalytic activity of 1a (0.1 mol %) preactivated with a less amount of 2b was far superior to that of the Bergens’s catalyst27 (TON = 970 with 1 mol % of 2b vs. 230 with 5 mol % of KN[Si(CH3)3]2 under the identical conditions (P H2 = 5 MPa, T = 100 °C, t = 24 h), giving 6h in 97% yield (Supplementary Table 1 for details). Products 6h and 6i are synthetic precursors of N, N-dimethyl-6-amino-1-hexanol, a polymerization initiator in polyurethane synthesis49. In marked contrast, when co-additives, L-Selectride (lithium tri-sec-butylborohydride) and NaB(C6H5)4, were used instead of NaH, the selective C–N bond cleavage was switched entirely to C=O bond cleavage, giving azepane in a quantitative yield (Fig. 3b). The hydrogenation of 3h to azepane proceeded under much milder conditions (P H2 = 2 MPa), compared with a very recent example in which catalytic (triphos)Ru derivatives were used (P H2 = ca. 10 MPa)14. Although 3h and inter-h might be in an equilibrium even under H2 40, the net reaction would proceed via either one of (or both of) the following two pathways involving the same reaction intermediate, N, O-hemiacetal inter-h, which undergoes (Fig. 3c): (1) direct cleavage of the C–O bond and subsequent hydrogenation of the resulting imine-h; or (2) the C–N bond cleavage that leads to the equilibrium (hydrogenation of the aldehyde ⇆ dehydrogenation of the resulting CH–OH group ⇆ intramolecular cyclization to inter-h), followed by the reaction steps shown in (1). Indeed, when aminoalcohol 6h was used instead of 3h, the identical hydrogenation conditions gave azepane almost quantitatively.

Hydrogenation of aromatic and cyclic amides. Unless otherwise specified, the reaction was performed using 1a (1 mol %), 3 (100 mol%), and NaH (6–10 mol %) in toluene. Pressure indicated is of H2 at 25 °C. nd: not determined. See Supplementary Information for experimental details. (a) Hydrogenation with P H2 = 0.5–3 MPa, T = 80–130 °C. (b) Additive effects on selective C=O bond cleavage in the hydrogenation of a cyclic amide and in the dehydrogenation/hydrogenation of a linear amino alcohol. (c) Possible reaction pathways starting from 3h and 6h under hydrogenation conditions. † 1a was preactivated: P H2(pre) = 1 MPa, T pre = 160 °C, t pre = 5 h.

Hydrogenation of various unactivated amides. Unless otherwise specified, the reaction was performed using 1a (1 mol %), 3 (100 mol%), and NaH (6–10 mol %) in toluene. Pressure indicated is of H2 at 25 °C. nd: not determined. See Supplementary Information for experimental details. (a) Comparison of effects of steric bulkiness of 3 on reaction conditions. † 1a was preactivated: P H2(pre) = 1 MPa, T pre = 160 °C, t pre = 5 h. (b) Hydrogenation of CO2-derived 3. ‡1 mol % 1b preactivated was used instead of 1a: P H2(pre) = 8 MPa, T pre = 160 °C, t pre = 2 h.
Hydrogenation of the more sterically demanding amides 3k, 3m, and 3n also took place, capitalizing on the structural robustness (negligible ligand detachment) of the catalyst even under harsher reaction conditions (P H2 = 3–8 MPa, T = 130–160 °C) (Fig. 4a). The mercury test38 was also employed, in which Hg(0) (150 mol %) was added during the hydrogenation of 3n to probe the possibility of catalysis by a Ru nanoparticle under the harshest conditions (P H2 = 8 MPa, T = 160 °C, t = 96 h). The catalytic activity was not perturbed during the course of the reaction (4n: 71%; 5f: 92%). This is in good contrast to previous results using the less stable precatalyst 2a 38, in which only marginal hydrogenation of 3m and 3n took place. In general, the more sterically demanding the amide, the less reactive the amide.
The hydrogenation rate of urea 3p with 1b was comparable to that obtained with 1a, while urethane derivative 3q was hydrogenated more effectively using 1b than 1a (Fig. 4b). Methanol was produced in ca. 60–70% yield in both cases, along with 4a and 5a in almost quantitative yields. These reactions are vitally important to the methanol economy8, 10, 11, 23, 24, as urea specifically is an excellent chemical reservoir and carrier of CO2.
Compared with 3p and 3q, hydrogenation of another CO2 derivative, DMF, proceeded far more smoothly, giving full conversion at 60 °C with P H2 = 8 MPa (Fig. 5a), and at 120 °C with P H2 = 2 MPa, producing CH3OH in ca. 60% yield in both cases. To hydrogenate tertiary amides DMF, 3j and 3k, and acetamide 3g, preactivation of 1a was required (P H2(pre) = 1 MPa, T pre = 160 °C, t pre = 5 h) before addition of the corresponding 3 to the catalyst mixture. Since the chemical immobilization of CO2 as DMF9 and hydrogenation of DMF10, 31 have well been investigated in addition to the present result, a combination of the previous and present methods could provide an alternative route that benefits the methanol economy at low T and/or P H2, in a future effort to improve the method of recovering/recycling Me2NH.

Multifaceted features of hydrogenation of small and highly functionalized amides. (a) DMF hydrogenation (b), Oligoamide hydrogenation. (c) Hydrogenation of 3u and 3v bearing directing groups. † 1a was preactivated: P H2(pre) = 1 MPa, T pre = 160 °C, t pre = 5 h. (d) Chemoselective hydrogenation of 3w (3w:4x:4y = 100:50:50 mol %). See Supplementary S63 for experimental details.
To the best of our knowledge, the selective and stepwise cleavage of the different C–N bonds in oligoamides such as diamide 3s and triamide 3t (a dipeptide with protection at the N-terminus) was successfully accomplished for the first time (Fig. 5b). The chiral centers epimerized, giving a racemic mixture of β-amino alcohols 6s and 6t. More intricately functionalized, commercial polyamides available from Toray Co. (AQ nylon P-70 and T-70 (105–120 mg each)) were also hydrogenated (1a, 2.9 mg; P H2 = 8 MPa, T = 160 °C, t = 48 h), giving different monomer units, where the mass balance before and after the reaction was ca. 80% consistent (the structures of the two monomer units cannot be disclosed here due to a confidentiality agreement with Toray). This might pave a new avenue for recycling monomer derivatives, as an alternative to the depolymerization of polyamides (e.g., 6-nylon) that proceeded at 300 °C50.
Finally, the synthetic potential, including the chemoselectivity, of the hydrogenation was investigated. A directing group of C–H bond functionalization (8-aminoquinoline)6 in 3u and of a catalytic amide aldol reaction (5v)7 in 3v were easily detached from a main alkyl chain through the catalytic C–N bond cleavage by H2 (Fig. 5c). The partial structure of 8-aminoquinoline (pyridine) was concomitantly hydrogenated, giving 5u in quantitative yield; in contrast, the pyridine moiety of 5v also obtained quantitatively remained unreacted with H2. The hydrogenation method using 1a is proven to have potential for converting rather thermodynamically stable, directing group-incorporated products to synthetically more useful alcohols. 5u and 5v are recyclable, and the former would also be reusable upon exploration of its rearomatization. Although the directing groups could be expected to strongly coordinate with a transition metal, H2 reacts even more favorably with the Ru center by taking advantage of a sterically confined H2-adsorption site. A more activated amide, anilide 3w, was hydrogenated rapidly, giving near quantitative yields of 4a and aniline with 0.25 mol % of 1a and 6 mol % of NaH (P H2 = 0.5 MPa, T = 80 °C, t = 48 h; TON = ~400). Furthermore, the amide group of 3w was hydrogenated (P H2 = 1 MPa, T = 80 °C, t = 27 h) preferentially even in the presence of a tri- and di-substituted olefins 4x and 4y (Fig. 5d). Since olefins are more likely to be hydrogenated via an inner-sphere mechanism51 (through direct interaction of the olefin with a metal center), these results again justify an outer-sphere mechanism18,19,20,21, 26,27,28,29, 33, 34, 37, 38, 42,43,44 that operates specifically for the hydrogenation of amides in preference to olefins, at least under mild conditions.
In conclusion, the effectiveness of a versatile “sterically confined bipyridine-Ru framework” for the C–N bond and C=O bond cleavage via hydrogenation of a variety of amides (from DMF to polyamides including diamides, triamides, and the synthetic polymers) under both mild and harsh reaction conditions has been demonstrated. Chemoselective amide hydrogenation, as well as hydrogenation of compounds potentially useful for the “methanol economy” under mild conditions, was also accomplished. Bipyridine (bpy) bearing two CH2 groups, two phosphorus atoms with sterically bulky alkyl substituents, and Ru are the unique constituents of the H2-adsorption site. These groups and elements are cooperative (to facilitate dearomatization and hydrogenation of bpy through deprotonation of the CH2 groups) and crucial for inducing robust ((P,(N, N)bpy, P)Ru structure which is electronically and sterically stabilized) and diverse (structures derived by different degrees of partial or full hydrogenation of bpy) catalysts. Further improvement of the robust (pre)catalyst based on our concept, including adopting a coordinatively-saturated Ru center, may significantly benefit the development of a better-performing catalyst for amide hydrogenation producing nonstandard peptides of pharmaceutically great importance, and even for facilitating a general method for the selective C=O bond cleavage of amide bonds.
Methods
Typical hydrogenation procedure
Under a continuous Ar flow, RUPIP2 (1a) (2.94 mg, 0.005 mmol), NaH (55% oil dispersion, 1.31 mg, 0.03 mmol), anhydrous toluene (1.5 mL), N-benzylbenzamide (3a) (105.6 mg, 0.5 mmol) and a magnetic stirring bar were placed in a dried Teflon tube (21 mL capacity). The Teflon tube was quickly inserted into an autoclave, and the autoclave inside was purged 10 times with hydrogen gas (1 MPa). The autoclave was pressurized with a 1 MPa of hydrogen gas at 25 °C, and heated at 110 °C for 24 h under stirring (800 rpm). The autoclave was cooled to ~25 °C in an ice–water bath, and the reaction mixture was quenched with a 2.0 M Et2O solution of HCl (15 μL, 0.03 mmol). The organic phase was removed in vacuo (ca. 100 mmHg, 40 °C). The residue was diluted with CDCl3, and analyzed by 1H NMR. The yields of benzyl alcohol (4a) (81%) and benzylamine (5a) (81%) were calculated based on the integral ratio among the signals of these compounds with respected to an internal standard (1,1,2,2-tetrachloroethane).
References
Smith, A. M. & Whyman, R. Review of methods for the catalytic hydrogenation of carboxamides. Chem. Rev. 114, 5477–5510, doi:10.1021/cr400609m (2014).
Werkmeister, S., Junge, K. & Beller, M. Catalytic hydrogenation of carboxylic acid esters, amides, and nitriles with homogeneous catalysts. Org. Process Res. Dev. 18, 289–302, doi:10.1021/op4003278 (2014).
Dub, P. A. & Ikariya, T. Catalytic reductive transformations of carboxylic and carbonic acid derivatives using molecular hydrogen. ACS Catal. 2, 1718–1741, doi:10.1021/cs300341g (2012).
He, G., Zhao, Y., Zhang, S., Lu, C. & Chen, G. Highly efficient syntheses of azetidines, pyrrolidines, and indolines via palladium catalyzed intramolecular amination of C(sp3)–H and C(sp2)–H bonds at γ and δ positions. J. Am. Chem. Soc. 134, 3–6, doi:10.1021/ja210660g (2012).
Nadres, E. T. & Daugulis, O. Heterocycle synthesis via direct C–H/N–H coupling. J. Am. Chem. Soc. 134, 7–10, doi:10.1021/ja210959p (2012).
Ano, Y., Tobisu, M. & Chatani, N. Palladium-catalyzed direct ethynylation of C(sp3)–H bonds in aliphatic carboxylic acid derivatives. J. Am. Chem. Soc. 133, 12984–12986, doi:10.1021/ja206002m (2011).
Weidner, K., Kumagai, N. & Shibasaki, M. A designed amide as an aldol donor in the direct catalytic asymmetric aldol reaction. Angew. Chem. Int. Ed. 53, 6150–6154, doi:10.1002/anie.201403118 (2014).
Olah, G. A., Prakash, G. K. S. & Goeppert, A. Anthropogenic chemical carbon cycle for a sustainable future. J. Am. Chem. Soc. 133, 12881–12898, doi:10.1021/ja202642y (2011).
Jessop, P. G., Hsiao, Y., Ikariya, T. & Noyori, R. Homogeneous catalysis in supercritical fluids: hydrogenation of supercritical carbon dioxide to formic acid, alkyl formates, and formamides. J. Am. Chem. Soc. 118, 344–355, doi:10.1021/ja953097b (1996).
Rezayee, N. M., Huff, C. A. & Sanford, M. S. Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 137, 1028–1031, doi:10.1021/ja511329m (2016).
Khusnutdinova, J. R., Gard, J. A. & Milstein, D. Combining low-pressure CO2 capture and hydrogenation to form methanol. ACS Catal. 5, 2416–2422, doi:10.1021/acscatal.5b00194 (2015).
Núñez Magro, A. A., Eastham, G. R. & Cole-Hamilton, D. J. The synthesis of amines by the homogeneous hydrogenation of secondary and primary amides. Chem. Commun. 3154–3156, doi:10.1039/b706635j (2007).
Coetzee, J. et al. Homogeneous catalytic hydrogenation of amides to amines. Chem. Eur. J. 19, 11039–11050, doi:10.1002/chem.201204270 (2013).
Meuresch, M., Westhues, S., Leitner, W. & Klankermayer, J. Tailor-made ruthenium-triphos catalysts for the selective homogeneous hydrogenation of lactams. Angew. Chem. Int. Ed. 55, 1392–1395, doi:10.1002/anie.201509650 (2016).
Stein, T. v. et al. Highly versatile catalytic hydrogenation of carboxylic and carbonic acid derivatives using a Ru-triphos complex: molecular control over selectivity and substrate scope. J. Am. Chem. Soc. 136, 13217–13225, doi:10.1021/ja506023f (2014).
Cabrero-Antonino, J. R., Alberico, E., Junge, K., Jungea, H. & Beller, M. Towards a general ruthenium-catalyzed hydrogenation of secondary and tertiary amides to amines. Chem Sci. 7, 3432–3442, doi:10.1039/C5SC04671H (2016).
Yuan, M.-L., Xie, J.-H., Zhu, S.-F. & Zhou, Q.-L. Deoxygenative hydrogenation of amides catalyzed by a well-defined iridium pincer complex. ACS Catal. 6, 3665–3669, doi:10.1021/acscatal.6b01019 (2016).
Ito, M., Sakaguchi, A., Kobayashi, C. & Ikariya, T. Chemoselective hydrogenation of imides catalyzed by Cp*Ru(PN) complexes and its application to the asymmetric synthesis of paroxetine. J. Am. Chem. Soc. 129, 290–291, doi:10.1021/ja067777y (2007).
Ito, M. et al. Hydrogenation of N-acylcarbamates and N-acylsulfonamides catalyzed by a bifunctional [Cp*Ru(PN)] complex. Angew. Chem. Int. Ed. 48, 1324–1327, doi:10.1002/anie.200805307 (2009).
Ito, M., Kobayashi, C., Himizu, A. & Ikariya, T. Highly enantioselective hydrogenative desymmetrization of bicyclic imides leading to multiply functionalized chiral cyclic compounds. J. Am. Chem. Soc. 132, 11414–11415, doi:10.1021/ja105048c (2010).
Ito, M. et al. Catalytic hydrogenation of carboxamides and esters by well-defined Cp*Ru complexes bearing a protic amine ligand. J. Am. Chem. Soc. 133, 4240–4242, doi:10.1021/ja1117254 (2011).
Balaraman, E., Gnanaprakasam, B., Shimon, L. J. W. & Milstein, D. Direct hydrogenation of amides to alcohols and amines under mild conditions. J. Am. Chem. Soc. 132, 16756–16758, doi:10.1021/ja1080019 (2010).
Balaraman, E., Gunanathan, C., Zhang, J., Shimon, L. J. W. & Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 3, 609–614, doi:10.1038/nchem.1089 (2011).
Balaraman, E., Ben-David, Y. & Milstein, D. Unprecedented catalytic hydrogenation of urea derivatives to amines and methanol. Angew. Chem. Int. Ed. 50, 11702–11705, doi:10.1002/anie.201106612 (2011).
Barrios-Francisco, R. et al. PNN ruthenium pincer complexes based on phosphinated 2,2′-dipyridinemethane and 2,2′-oxobispyridine. metal–ligand cooperation in cyclometalation and catalysis. Organometallics 32, 2973–2982, doi:10.1021/om400194w (2013).
Takebayashi, S., John, J. M. & Bergens, S. H. Desymmetrization of meso-cyclic imides via enantioselective monohydrogenation. J. Am. Chem. Soc. 132, 12832–12834, doi:10.1021/ja105783u (2010).
John, J. M. & Bergens, S. H. A highly active catalyst for the hydrogenation of amides to alcohols and amines. Angew. Chem. Int. Ed. 50, 10377–10380, doi:10.1002/anie.201103137 (2011).
John, J. M., Loorthuraja, R., Antoniuk, E. & Bergens, S. H. Catalytic hydrogenation of functionalized amides under basic and neutral conditions. Catal. Sci. Technol. 5, 1181–1186, doi:10.1039/C4CY01227E (2015).
Cabrero-Antonino, J. R. et al. Efficient base-free hydrogenation of amides to alcohols and amines catalyzed by well-defined pincer imidazolyl–ruthenium complexes. ACS Catal. 6, 47–54, doi:10.1021/acscatal.5b01955 (2016).
Zhang, J., Leitus, G., Ben-David, Y. & Milstein, D. Efficient homogeneous catalytic hydrogenation of esters to alcohols. Angew. Chem. Int. Ed. 45, 1113–1115, doi:10.1002/anie.200503771 (2006).
Rezayee, N. M., Samblanet, D. C. & Sanford, M. S. Iron-catalyzed hydrogenation of amides to alcohols and amines. ACS Catal. 6, 6377–6383, doi:10.1021/acscatal.6b01454 (2016).
Schneck, F., Assmann, M., Balmer, M., Harms, K. & Langer, R. Selective hydrogenation of amides to amines and alcohols catalyzed by improved iron pincer complexes. Organometallics 35, 1931–1943, doi:10.1021/acs.organomet.6b00251 (2016).
Kuriyama, W., Ino, Y., Ogata, O., Sayo, N. & Saito, T. A Homogeneous catalyst for reduction of optically active esters to the corresponding chiral alcohols without loss of optical purities. Adv. Synth. Catal. 352, 92–96, doi:10.1002/adsc.200900114 (2010).
Kuriyama, W. et al. Catalytic hydrogenation of esters. Development of an efficient catalyst and processes for synthesising (R)-1,2-propanediol and 2-(l-menthoxy)ethanol. Org. Process Res. Dev. 16, 166–171, doi:10.1021/op200234j (2016).
Garg, J. A., Chakraborty, S., Ben-David, Y. & Milstein, D. Unprecedented iron-catalyzed selective hydrogenation of activated amides to amines and alcohols. Chem. Commun. 52, 5285–5288, doi:10.1039/c6cc01505k (2016).
Shi, L. et al. Direct catalytic hydrogenation of simple amides: a highly efficient approach from amides to amines and alcohols. Chem. Eur. J. 23, 546–548, doi:10.1002/chem.201604904 (2017).
Kita, Y., Higuchi, T. & Mashima, K. Hydrogenation of amides catalyzed by a combined catalytic system of a Ru complex with a zinc salt. Chem. Commun. 50, 11211–11213, doi:10.1039/c4cc04481a (2014).
Miura, T., Held, I. E., Oishi, S., Naruto, M. & Saito, S. Catalytic hydrogenation of unactivated amides enabled by hydrogenation of catalyst precursor. Tetrahedron Lett. 54, 2674–2678, doi:10.1016/j.tetlet.2013.03.047 (2013).
Iida, K., Miura, T., Ando, J. & Saito, S. The dual role of ruthenium and alkali base catalysts in enabling a conceptually new shortcut to N‑unsubstituted pyrroles through unmasked α-amino aldehydes. Org. Lett. 15, 1436–1439, doi:10.1021/ol4001262 (2013).
Takada, Y., Iida, M., Iida, K., Miura, T. & Saito, S. Versatile ruthenium complex “RuPCY” for directed catalytic hydrogen management in organic synthesis. J. Org. Syn. Chem. Jpn. 74, 1078–1089, doi:10.5059/yukigoseikyokaishi.74.1078 (2016).
Saito, S. et al. Preliminary results of this report (including Ru complexes 1a–e) were used in application for a patent: JP patent, Appl. #2013–42385, Filed: Mar 4 (2013).
Sandoval, C. A., Ohkuma, T., Muñiz, K. & Noyori, R. Mechanism of asymmetric hydrogenation of ketones catalyzed by BINAP/1,2-diamine-ruthenium(II) complexes. J. Am. Chem. Soc. 125, 13490–13503, doi:10.1021/ja030272c (2003).
Noyori, R. & Ohkuma, T. Asymmetric catalysis by architectural and functional molecular engineering: practical chemo- and stereoselective hydrogenation of ketones. Angew. Chem. Int. Ed. 40, 40–73, doi:10.1002/1521-3773(20010105)40:1<40::AID-ANIE40>3.0.CO;2-5 (2001).
Yamakawa, M., Ito, H. & Noyori, R. The metal–ligand bifunctional catalysis: a theoretical study on the ruthenium(II)-catalyzed hydrogen transfer between alcohols and carbonyl compounds. J. Am. Chem. Soc. 122, 1266–1478, doi:10.1021/ja991638h (2000).
Li, W., Xie, J.-H., Yuan, M.-L. & Zhou, Q.-L. Ruthenium complexes of tetradentate bipyridine ligands: highly efficient catalysts for the hydrogenation of carboxylic esters and lactones. Green Chem. 16, 4081–4085, doi:10.1039/C4GC00835A (2014).
Langer, R. et al. Stepwise metal–ligand cooperation by a reversible aromatization/deconjugation sequence in ruthenium complexes with a tetradentate phenanthroline-based ligand. Chem. Eur. J 19, 3407–3414, doi:10.1002/chem.201204003 (2013).
Sergeev, A. G. & Hartwig, J. F. Selective, nickel-catalyzed hydrogenolysis of aryl ethers. Science 332, 439–443, doi:10.1126/science.1200437 (2011).
Garrou, P. E. Transition-metal-mediated phosphorus-carbon bond cleavage and its relevance to homogeneous catalyst deactivation. Chem. Rev. 85, 171–185, doi:10.1021/cr00067a001 (1985).
Imabeppu, M., Kiyoga, K., Okamura, S., Shoho, H. & Kimura, H. One-step amination of α,ω-alkylenediols over Cu/Ni-based catalysts. Catal. Commun. 10, 753–757, doi:10.1016/j.catcom.2008.10.046 (2009).
Kamimura, A. & Yamamoto, S. An efficient method to depolymerize polyamide plastics: A new use of ionic liquids. Org. Lett. 9, 2533–2535, doi:10.1021/ol070886c (2007).
Ohkuma, T., Ooka, H., Ikariya, T. & Noyori, R. Preferential hydrogenation of aldehydes and ketones. J. Am. Chem. Soc. 117, 10417–10418, doi:10.1021/ja00146a041 (1995).
Acknowledgements
This work was supported by funds from Asahi Glass Foundation, scientific research on innovation areas “Molecular Activation Directed toward Straightforward Synthesis (Grant #25105721)”, “Precisely Designed Catalysts with Customized Scaffolding, (Grant #16H01012)”, MEXT, Japan, and “Advanced Catalytic Transformation program for Carbon utilization (ACT-C, Grant #JPMJCR12YJ)”, JST, Japan. T.M. & M.N. acknowledges JSPS fellowship (to T.M.), as well as IGER (NU) SRA, Iue Memorial, and Iwadare fellowships (to M.N.). The authors wish to thank Profs. J. F. Hartwig (UC Berkeley) and R. Noyori (NU & RIKEN) for fruitful discussions. We are also grateful to K. Oyama and Y. Maeda (Chemical Instrument Room, NU) for technical support. We dedicate this manuscript to Professor Takao Ikariya with the deepest condolences on the occasion of his sad death.
Author information
Authors and Affiliations
Contributions
T.M. made the initial discovery, and carried out main experiments. M.N. synthesized ruthenium complexes and checked the reproducibility of synthetic and analytical experiments. K.T. and T.S. contributed equally to carry out parts of experiments. S.S. directed the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miura, T., Naruto, M., Toda, K. et al. Multifaceted catalytic hydrogenation of amides via diverse activation of a sterically confined bipyridine–ruthenium framework. Sci Rep 7, 1586 (2017). https://doi.org/10.1038/s41598-017-01645-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01645-z
This article is cited by
-
Chemoselectivity change in catalytic hydrogenolysis enabling urea-reduction to formamide/amine over more reactive carbonyl compounds
Nature Communications (2023)
-
Recent global insight into mitigation of plastic pollutants, sustainable biodegradable alternatives, and recycling strategies
International Journal of Environmental Science and Technology (2023)
-
Catalytic transformation of functionalized carboxylic acids using multifunctional rhenium complexes
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
