
Abstract
Despite the rapid advances in sequencing technology, limited genomic resources are currently available for phytophagous spider mites, which include many important agricultural pests. One of these pests is Tetranychus piercei (McGregor), a serious banana pest in East Asia exhibiting remarkable tolerance to high temperature. In this study, we assembled a high-quality genome of T. piercei using a combination of PacBio long reads and Illumina short reads sequencing. With the assistance of chromatin conformation capture technology, 99.9% of the contigs were anchored into three pseudochromosomes with a total size of 86.02 Mb. Repetitive elements, accounting for 14.16% of this genome (12.20 Mb), are predominantly composed of long-terminal repeats (30.7%). By combining evidence of ab initio prediction, transcripts, and homologous proteins, we annotated 11,881 protein-coding genes. Both the genome and proteins have high BUSCO completeness scores (>94%). This high-quality genome, along with reliable annotation, provides a valuable resource for investigating the high-temperature tolerance of this species and exploring the genomic basis that underlies the host range evolution of spider mites.
Similar content being viewed by others


Background & Summary
Phytophagous spider mites in Tetranychidae comprise more than 1,300 species, many of which are serious agricultural pests, such as Tetranychus urticae Koch, Panonychus citri McGregor1. Spider mites are notorious for developing rapid resistance to pesticides, causing significant economic losses since the widespread use of synthetic insecticides and fungicides after World War II2. The global acaricide market was valued at up to 400 million dollars annually3. In addition to their economic importance, spider mites exhibit diverse host ranges, from monophagous (e.g. Tetranychus lintearius) to extremely polyphagous (e.g. T. urticae)4, making them an ideal system for exploring the mechanisms underlying the evolution of host range. The two-spotted spider mite (T. urticae) is the first species of Chelicerate to have its whole genome sequenced, which was obtained by Sanger sequencing and assembled at the scaffold level5. Subsequently, in 2019, the genome was further refined and anchored into three pseudochromosomes6. This genome provides important insights into the host adaptation and pesticide resistance evolution of T. urticae and suggests a possible link between its rapid development of pesticide resistance and its strong adaptive ability to host plants5,7.
Tetranychus piercei (McGregor) is a major pest on banana (Musa spp.), papaya (Carica papaya) and other crops in East Asia8,9,10. It can also feed on plants such as mulberry (Morus alba), rose (Rosa sp.), peach (Prunus persica), sweetsop (Annona squamosa), cassava (Manihot esculenta) and fig (Ficus carica)4. Being the phylogenetically older sister species of T. urticae, T. piercei has a narrower host range and distinct host plant preferences11,12. Notably, T. piercei exhibits greater tolerance to high temperature compared to T. urticae, positioning it as a potential replacement for T. urticae as a major pest in the context of global warming13. However, limited information on its genetic resources hinders our understanding of its strong tolerance to high temperature and the evolution of the detoxification system in Tetranychinae.
In this study, we employed a combination of PacBio continuous long reads, accurate Illumina short reads and chromosomal conformation capture (Hi-C) data to assemble a chromosomal-level genome of T. piercei, which includes 3 chromosomes and 11,881 protein-coding genes. Synteny analysis revealed dramatic chromosomal rearrangement between T. piercei and T. urticae. This high-quality genome will facilitate in-depth biological studies of T. piercei and enable exploration of the genomic basis underlying the host range evolution of spider mites.
Methods
Raw material collection
At least 100 wild spider mites, including larvae, nymphs, and adults, were collected from Trachycarpus fortunei in Sanya Hainan province (18.29°N, 109.47°E), a tropical region in China. We amplified the nuclear ribosomal internal transcribed spacer (ITS) sequence to confirm the species identification of T. piercei14. By backcrossing male sons with a virgin female, an isofemale line was constructed to minimize heterozygosity. The isofemale line was reared on potted soybeans at a population size of thousands for at least 20 generations.
DNA and RNA sequencing
Total genomic DNA was extracted from more than 5,000 adult females using MagAttract® HMW DNA Kit. The PacBio 30 kb SMRTbell library was prepared with more than 5 μg gDNA using the SMRTbellTM Prep Kit 2.0 (Pacific Biosciences). The mode of Continuous Long Read (CLR) was run on the Sequel IIe system, and generated 6.44 Gbp raw data (75-fold depth). Illumina whole-genome sequencing was prepared using a 350 bp-insert fragment library (150 bp paired-end) by Truseq DNA PCR-free Kit, which was further sequenced on an Illumina NovaSeq 6000 platform. High-throughput chromosome conformation capture (Hi-C) included cross-linking, HindIII restriction enzyme digestion, end repair, DNA cyclization, purification and capture. The Hi-C library with 300–700 bp insert size library was sequenced on the NovaSeq 6000 platform, and 8.45 Gbp reads were generated to scaffold chromosomes. Total RNA was extracted from 100 adult females feeding on common beans using TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s instructions. RNA library was constructed using the VAHTS mRNA-seq v2 Library Prep Kit (Vazyme, Nanjing, China) and sequenced on an Illumina NovaSeq 6000 platform. Finally, we generated 6.44 Gb (75×) PacBio long reads, 13.10 Gb (152×) Illumina short reads, 8.45 Gb Hi-C (98×) reads, and 12.62 Gb transcriptome reads for our genome assembly.
Genome survey
Duplicate and low-quality Illumina raw reads (base quality < Q20, length < 15 bp, polymer A/G/C > 10 bp) were trimmed and removed using BBtools package v38.8215. The 21-mer depth distribution was counted using script khist.sh of BBtools v38.82. GenomeScope v2.016 was used to estimate the genome size and heterozygosity of T. piercei with the maximum kmer coverage at 1,000×. Based on the distribution of kmer coverage and frequency, the estimated genome size of T. piercei was 86.45 Mb, with a heterozygosity rate of around 0.001% and a repeat content proportion of approximately 14.6% (Fig. 1).

Genome survey at 21-mer of T. piercei estimated by GenomeScope. The vertical dotted lines represent the peaks of different coverages for the heterozygous, the homozygous, and the duplicated sequences (the last two peaks) separately.
Chromosome staining
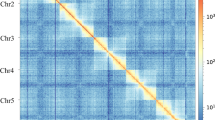
Unfertilized (haploid) and fertilized (diploid) eggs of T. piercei, laid within 8 hours, were collected in a centrifuge tube containing phosphate buffered saline (PBS; 0.85% NaCl, 1.4 mM KH2PO4, 8 mM Na2HPO4, PH 7.1). The PBS was then discarded, and 500 μL 50% sodium hypochlorite solution was added, allowing it to stand for 2–3 minutes. After discarding the sodium hypochlorite solution, a mixture of hexane and methanol (1:1) was added to the centrifuge tube, and the contents were vigorously shaken for 3 min to remove the chorion. The eggs were rehydrated through an ethanol series (95, 70, 50 and 35%) and then washed in PBT (PBS containing 0.1% Triton X-100) for 15 min. After being washed five times for 1 min each in PBS, the eggs were incubated with a fluorescence quenching agent containing DAPI at room temperature for 5 min. Subsequently, the eggs were mounted on slides and covered with coverslips for further microscopic investigation using a Leica TCS SP8 confocal microscope. The egg DNA staining of diploid females and haploid males (Fig. 2a,b) consistently indicates that the genome of T. piercei consists of three chromosomes.

Chromosome staining, Hi-C heatmap and synteny. (a) DAPI staining of chromosomes in diploid female egg of T. piercei. The blue signals represent condensed chromosomal regions, while the red dashed lines in the model panel represent simulated chromosome boundaries. (b) DAPI staining of chromosomes in haploid male egg. (c) Genome-wide chromosomal interaction heatmap generated in Hi-C interaction analysis with each chromosome in the blue box. The frequency of Hi-C interaction links is represented by the color, which ranges from white (low) to red (high). (d) Synteny dot plot based on protein homologous between T. piercei and T. urticae.
Genome assembly
The CLR reads were set as input to Flye v2.9 and Raven v1.6.0 to assemble continuous long reads17,18. The better assembly from Flye, which had a greater N50 length and completeness, was retained as the following primary assembly. One round of built-in long reads polishing was performed by Flye v2.9. Then, two rounds of short reads were used to polish and fill in gaps of the primary assembly with NextPolish v1.4.019. Haplotigs and duplication caused by haplotype divergence were eliminated by Purge_dups v1.2.5 using the alignment program Minimap2 v2.2320,21. Hi-C reads were aligned to the purged genome using BWA v0.7.1722 to anchor, order and orient contigs into chromosomal assembly following 3D-DNA pipeline23. Then, we manually reviewed and corrected assembled errors using Juicebox v1.11.0824. Vector contaminants were checked against the UniVec database using BLAST + v2.11.0 with the VecScreen parameters25. Bacterial and human being contaminants were detected in the assembly against the Nt database using MMseqs v13-4511126. The completeness of genome assembly was evaluated by BUSCO version 5.2.227 using the arachnida_odb10 dataset (creation date 2020-08-05). The reads from the whole genome sequencing were aligned back to the genome assembly to access the mapping rate.
After de novo assembly, polishing and purging, 56 contigs (N50 of 3.33 Mb) with a total length of 86.13 Mb were generated, accounting for 99.63% of the estimated genome. By combining Hi-C with high-throughput sequencing and manual adjustment, we anchored these contigs into 4 scaffolds, including 3 megascaffolds and 1 unplaced scaffold (Fig. 2c). This unplaced scaffold was identified as bacterial contamination by NCBI and was subsequently excluded. Finally, a total length of 86.02 Mb contigs was assigned to 3 pseudochromosomes (Fig. 3), with scaffold N50 of 29.25 Mb (Table 1). The GC content of the T. piercei genome was 32.17%, which is similar to that of T. urticae (32.25%).

Circular karyotype representation of the chromosomes in non-overlapping windows of 100 kb. Tracks from inside to outside are GC content, gene density, Gypsy density, Copia density and DNA transposons density.
Genome annotation
The repetitive elements were identified using RepeatModeler v2.0.2, which discovered the complete long terminal repeats (LTR) with the integration of LTRharvest and LTR_retriever28. RepeatMasker v4.1.2p1 and RMBlast v2.11.0 were searched against the custom repeat library of Dfam 3.5 and Repbase v20181026 to soft mask repeats of the genome assembly29,30,31. Two ab initio gene prediction software, BRAKER2 v2.1.6 and GeMoMa v1.832,33, were used to find protein-coding gene structure based on the masked genome. Transcriptome mapping conducted by HISAT2 v2.2.0 and homologous proteins from five species (Daphnia magna, Dermacentor silvarum, Drosophila melanogaster, T. urticae, and Varroa destructor) were provided to assist gene prediction of BRAKER2/GeMoMa. Genome-guided transcript assembly was performed by StringTie v2.1.6, and the results were used as mRNA evidence for MAKER2 v3.01.0334,35. The same homologous proteins were also fed to MAKER2 as the protein evidence. Finally, MAKER2 combined ab initio prediction, mRNA and homology-protein evidence to generate gene models with direct predictions not allowed for transcripts and proteins. Proteins with lengths shorter than 30 aa were discarded. The functional annotation (Table S1) of predicted protein sequences was searched against UniProt, InterProScan and eggNOG databases. Diamond v2.0.1136 under the ‘very sensitive’ mode was used to assign gene function of the best hits in the UniProt database. Protein domains, Gene Ontology terms and pathways were supported by Pfam, SMART, Superfamily and CDD using InterProScan537. Complementary annotation of ko, KEGG categories were provided using eggNOG-mapper v2.1.5 against EggNOG 5.0 database38,39.
To explore chromosomal synteny between T. piercer and T. uritcae6, we conducted a homologous search between their respective protein sequences using Diamond v2.0.1136 with default parameters. The resulting homologous dot plot was visualized to detect collinearity, inversions and translocations using WGDI v0.6.540.
Data Records
The raw reads and genome assembly have been deposited in the NCBI databases under BioProject PRJNA833563. The PacBio, Illumina, Hi-C, and transcriptome data are available under identification numbers SRR23622209-SRR2362221241. The final chromosome assembly has been deposited at GenBank under the accession number GCA_036759885.142. The final chromosome-level genome assembly, annotation, and protein sequences are available at the Figshare database (https://doi.org/10.6084/m9.figshare.22215145)43.
Technical Validation
Evaluation of the genome assembly
Compared to T. urticae6, our genome assembly exhibits greater contiguity and completeness attributed to the utilization of long reads and Hi-C sequencing. The contigs number, contigs N50 of T. piercei are much better than those of the two-spotted spider mite (56 vs. 1,996; 3.33 Mb vs. 0.21 Mb; Table 1). We mapped whole-genome resequencing reads to the T. piercei genome and found that 92.5% of PacBio long reads and 96.51% of Illumina short reads could be well aligned. The complete benchmarking universal single-copy orthologs (BUSCOs) under genome mode were used to assess the genome completeness. A total of 94.6% (2,775/2,934) complete BUSCOs were identified, including 89.0% (2,612) single-copy BUSCOs, 5.6% (163) duplicated BUSCOs, 0.7% (22) fragmented BUSCOs, and 4.7% (137) missing BUSCOs.
Repeat elements and protein-coding genes
A total of 12.20 Mb repetitive elements were identified, accounting for 14.16% of the genome (Fig. 3). Besides unclassified repeats (4.67%), long-terminal repeat (LTR) represented the most common repeat element (4.36%), followed by simple repeats (2.04%), DNA transposons (1.67%), long-interspersed elements (LINE, 0.68%), and others (0.75%).
Using multiple lines of evidence, we annotated 11,881 protein-coding genes for T. piercei (Table 1). Most genes (>97%) had ‘annotation edit distance’ (AED) scores smaller than 0.5, indicating strong support from the evidence of transcript and homologous protein. Under the protein model, the complete BUSCOs for our genome annotation were 2,773 (94.5%). Although T. urticae had more coding genes than T. piercei (19,102 vs. 11,881), the results of BUSCOs suggested that T. piercei had higher completeness (90.4% vs. 94.5%; Table 1). Based on the synteny of homologous proteins, we found that the chromosomes of the two species underwent dramatic inversion and translocation events (Fig. 2d). More than half of Chr3 in T. piercer is syntenic to fragments of Chr1 and Chr2 in T. urticae.
Code availability
All commands and pipelines were executed following the manuals and protocols of the corresponding bioinformatic software. The versions and parameters of the software have been detailed in the Methods section.
References
Helle, W. & Sabelis, M. W. Spider Mites: Their Biology, Natural Enemies and Control. 1A (Elsevier, Amsterdam, 1985).
Walter, D. E. & Proctor, H. C. Mites: Ecology, Evolution and Behaviour. (Springer, Dordrecht, 2013).
Van Leeuwen, T., Tirry, L., Yamamoto, A., Nauen, R. & Dermauw, W. The economic importance of acaricides in the control of phytophagous mites and an update on recent acaricide mode of action research. Pestic. Biochem. Physiol. 121, 12–21 (2015).
Migeon, A., Nouguier, E. & Dorkeld, F. Spider Mites Web: a comprehensive database for the Tetranychidae. Trends in Acarology 557–560 (2010).
Grbić, M. et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 479, 487–492 (2011).
Wybouw, N. et al. Long-Term Population Studies Uncover the Genome Structure and Genetic Basis of Xenobiotic and Host Plant Adaptation in the Herbivore Tetranychus urticae. Genetics 211, 1409–1427 (2019).
Dermauw, W. et al. A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae. Proc. Natl. Acad. Sci. USA 110, E113–E122 (2013).
Fu, Y., Zhang, F., Peng, Z., Liu, K. & Jin, Q. The effects of temperature on the development and reproduction of Tectranychus Tetranychus piercei McGregor (Acari: Tetranychidae) in banana. Syst. Appl. Acarol. 7, 69 (2002).
Ullah, M. S., Gotoh, T. & Lim, U. T. Life history parameters of three phytophagous spider mites, Tetranychus piercei, T. truncatus and T. bambusae (Acari: Tetranychidae). J. Asia-Pac. Entomol. 17, 767–773 (2014).
Ohno, S. et al. Non-crop host plants of Tetranychus spider mites (Acari: Tetranychidae) in the field in Okinawa, Japan: Determination of possible sources of pest species and inference on the cause of peculiar mite fauna on crops. Appl. Entomol. Zool. 45, 465–475 (2010).
Hu, Q.-Q. et al. Phylogenetic-Related Divergence in Perceiving Suitable Host Plants among Five Spider Mites Species (Acari: Tetranychidae). Insects 13, 705 (2022).
Matsuda, T., Kozaki, T., Ishii, K. & Gotoh, T. Phylogeny of the spider mite sub-family Tetranychinae (Acari: Tetranychidae) inferred from RNA-Seq data. PLoS ONE 13, e0203136 (2018).
Gotoh, T., Moriya, D. & Nachman, G. Development and reproduction of five Tetranychus species (Acari: Tetranychidae): Do they all have the potential to become major pests? Exp. Appl. Acarol. 66, 453–479 (2015).
Ge, C., Ding, X.-L., Zhang, J.-P. & Hong, X.-Y. Tetranychus urticae (green form) on Gossypium hirsutum in China: two records confirmed by aedeagus morphology and RFLP analysis. Syst. Appl. Acarol. 18, 239–245 (2013).
Bushnell, B., Rood, J. & Singer, E. BBMerge – Accurate paired shotgun read merging via overlap. PLoS ONE 12, e0185056 (2017).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Vaser, R. & Šikić, M. Time- and memory-efficient genome assembly with Raven. Nat. Comput. Sci. 1, 332–336 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinforma. 25, (2009).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 3, lqaa108 (2021).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19, 189 (2018).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Sun, P. et al. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 15, 1841–1851 (2022).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP424604 (2023).
Sun, J.-T. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_036759885.1 (2024).
Chen, L., Zhang, F. & Sun, J.-T. The genome assembly and annotation of Tetranychus piercei, figshare, https://doi.org/10.6084/m9.figshare.22215145.v5 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. U2003112, 32020103011, 32202290) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20221003).
Author information
Authors and Affiliations
Contributions
J.T.S., F.Z. and L.C. designed the research. X.Y.Y. contributed to the sampling and sequencing. F.Z. assembled and annotated the genome. L.C., X.Y.Y., H.M.Z. and L.G.X. analyzed the data and drew the pictures. L.R. performed chromosome staining. L.C. and J.T.S. wrote the draft manuscript, and X.Y.H. improved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Yu, XY., Zhang, F. et al. A chromosome-level genome assembly of the spider mite Tetranychus piercei McGregor. Sci Data 11, 340 (2024). https://doi.org/10.1038/s41597-024-03189-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03189-0
