
Abstract
Trichoptera is one of the most evolutionarily successful aquatic insect lineages and is highly valued value in adaptive evolution research. This study presents the chromosome-level genome assemblies of Himalopsyche anomala and Eubasilissa splendida achieved using PacBio, Illumina, and Hi-C sequencing. For H. anomala and E. splendida, assembly sizes were 663.43 and 859.28 Mb, with scaffold N50 lengths of 28.44 and 31.17 Mb, respectively. In H. anomala and E. splendida, we anchored 24 and 29 pseudochromosomes, and identified 11,469 and 10,554 protein-coding genes, respectively. The high-quality genomes of H. anomala and E. splendida provide critical genomic resources for understanding the evolution and ecology of Trichoptera and performing comparative genomics analyses.
Similar content being viewed by others


Background & Summary
Trichoptera, commonly known as caddisflies, represent the largest order of completely aquatic insects within Endopterygota1. Encompassing approximately 17,000 extant species, Trichoptera are distributed across all continents except Antarctica2. Their larvae exhibit remarkably diverse behavior, constructing various nest structures or living freely in aquatic environments3. Their adaptability to varying water conditions, including temperature and dissolved oxygen, differs significantly among families, genera, and individual species4. Consequently, they serve as vital indicator organisms in water quality monitoring efforts. Additionally, the varied feeding habits of trichopteran larvae contribute to the energy dynamics within stream ecosystems5,6.
Trichoptera is divided into two suborders, Annulipalpia and Integripalpia, based on morphology and habit. Annulipalpian larvae typically inhabit running water or wave-washed riverbanks, using pin silk along with plant debris and small stones to construct fixed shelter. Integripalpia includes “cocoon-makers” and “Phryganides”7,8. Cocoon-makers larvae are either free-living or construct purse-case or saddle-case and are usually found in fast-flowing rivers and streams. Last instar larvae produce closed, semipermeable cocoons for pupation. In contrast, most Phryganides larvae thrive in stagnant or slow-moving water, adeptly combining stones, leaves, and twigs with silk proteins to construct mobile nests9,10. Rhyacophilidae and Phryganeidae are representative cocoon-makers and Phryganides, respectively, and exhibit marked ecological habit and lifestyle differences.
The family Rhyacophilidae originated in the Palaearctic region and is primarily distributed in the northern-hemisphere11. Their predatory larvae exhibit high sensitivity to environmental changes12. However, the majority of phryganeid larvae are shredders, feeding on detritus and plant material in aquatic environments13. These larvae tend to be less sensitive to environmental changes compared with rhyacophilid larvae. Some species can survive in humid terrestrial environments after leaving the water10. Himalopsyche anomala Banks and Eubasilissa splendida Yang & Yang are typical representatives of Rhyacophilidae and Phryganeidae, respectively. Despite extensive studies on their biological characteristics, their precise phylogenetic positions and the molecular mechanisms underlying their adaptive evolution remain uncertain. High-quality reference genomes are crucial for advancing genetics and genome research. To date, nearly 30 trichopteran species have had their genomes sequenced and published, including two Himalopsyche species and Eubasilissa regina. However, the chromosome-level has been reached in only partial species from five families (Glossosomatidae, Hydropsychidae, Leptoceridae, Limnephilidae, and Odontoceridae).
To enhance our understanding of the adaptive evolution and ecology of holometabola aquatic insects, we used PacBio long-read sequencing, Illumina short-read sequencing, and Hi-C data sequencing techniques to achieve the first chromosome-level genome assemblies for H. anomala Banks and E. splendida Yang & Yang, with assembly sizes of 663.43 and 859.28 Mb and scaffold N50 lengths of 28.44 and 31.17 Mb, respectively. Hi-C scaffolding resulted in chromosome-level assemblies, with 99.29% (2,697 contigs) and 99.61% (643 contigs) of the initially assembled sequences anchored to 24 and 29 pseudochromosomes for H. anomala and E. splendida, respectively. In total, 288.10 Mb (43.43%) and 471.23 Mb (54.84%) of the sequences were identified as repetitive elements in these two respective assemblies. Moreover, integrating three prediction methods enabled the identification of 11,469 and 10,554 protein-coding genes (PCGs) in H. anomala and E. splendida, respectively. The high-quality genomes of these species not only advance our understanding of adaptive evolution in Trichoptera but also serve as resources for comparative genomics research on evolution in biology and ecology fields. Furthermore, they contribute to elucidating the phylogenetic relationships between the cocoon-maker and Phryganides groups.
Methods
Sample collection
Himalopsyche anomala and E. splendida specimens were collected using ultraviolet light tubes from Xi-niu Sea (33°11′42″N; 103°53′46″E; alt: 2,348 m) and Wu-hua Sea (33°09′32″N; 103°51′55″E; alt: 2,377 m), respectively, in Jiuzhaigou National Nature Reserve, Sichuan Province, in July 2020. Specimens were identified by X-Y Ge and C-H Sun. Each sample underwent cleaning with phosphate-buffered saline buffer and the gut was removed under a stereo microscope (to minimize intestinal microbial contamination). Subsequently, samples were stored in liquid nitrogen before nucleic acid extraction14.
Nucleic acid extraction and sequencing
For genome survey, transcriptome, PacBio, and Hi-C sequencing, four male individuals of each species were sequenced. Additionally, a female individual underwent DNA sequencing using the Illumina platform to identify sex chromosome. DNA and RNA were extracted from samples using the Qiagen DNeasy Blood & Tissue Kit (Qiagen) and TRIzol Reagent Kit (Invitrogen)15.
For PacBio sequencing, sequencing libraries with 20 kb (H. anomala) and 30 kb (E. splendida) insert size were constructed, respectively, using the SMRTbell Template Prep Kit 1.0-SPv3, tailored to the quality of extracted DNA. Long-read sequencing was performed using the PacBio Sequel II platform with the CLR strategy. PCR-free sequencing libraries with a 350 bp insert size were generated for short-read genome sequencing. The Hi-C library was created using Mbol restriction endonuclease16. Both library types were subsequently sequenced on the Illumina Novaseq. 6000 and BGISEQ-500 platforms.
In total, approximately 285.76 and 352.18 Gb of raw data were generated for H. anomala and E. splendida, respectively. For H. anomala, the raw data included 117.23 Gb (approximately 176×) of PacBio reads with a scaffold N50 of 19.78 kb, 86.45 Gb of Illumina reads (comprising 28.87 and 57.58 Gb from the female and male samples, respectively), 74.62 Gb of Hi-C data, and 6.11 Gb of transcriptome data. For E. splendida, the raw data consisted of 117.9 Gb (approximately 136×) of PacBio reads with a scaffold N50 of 29.33 kb, 131.42 Gb of Illumina reads (comprising 43.73 and 87.69 Gb from the female and male samples, respectively), 91.40 Gb of Hi-C data, and 6.16 Gb of transcriptome data.
Genome size estimation and assembly
The acquired DNA sequencing reads underwent rigorous quality control using BBmap v38.6717. This process included the removal of duplicate reads and filtering of low-quality reads, which were defined as follows: quality score < 20, length < 15, and consecutive polymer A/G/C > 10. For k-mer analysis, khist.sh was used with the parameter k = 21. Genome size was estimated using the R package of GenomeScope v2.0.118 to calculate the k-mer distribution and generate a histogram, with a maximum sequencing coverage of 10,000. The estimated genome sizes were approximately 608.17 and 786.73 Mb for H. anomala and E. splendida, respectively, with the H. anomala genome exhibiting higher heterozygosity (1.03%; Fig. S1) compared to the lower heterozygosity of E. splendida (0.79%; Fig. S2).
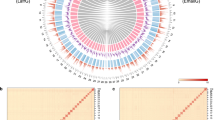
Flye v2.8.319 was used for PacBio long-read assembly, with one round of self-polishing based on long reads. This resulted in 774.15 and 870.01 Mb assemblies for H. anomala and E. splendida, respectively. Illumina short-read mapping was performed using Minimap2 v2.1720, and the assembled genome underwent two rounds of polishing with NextPolish v1.1.021. Redundant sequences were removed using Purge_Dups v1.2.522 with the haploid cutoff set at 60 (-s 60) based on the aforementioned short-read mapping. Before chromosome anchoring, Hi-C reads alignment and quality control were conducted using Juicer v1.6.223 with its default parameters. Subsequently, 3D-DNA v18092224 was employed to automatically anchor the majority of contigs into pseudochromosomes. Mis-joins were corrected using Juicebox v1.11.0823 through manual inspection and refinement. In total, 97.68% and 99.58% of assembly contigs were anchored into 24 and 29 pseudochromosomes, with lengths of 11.53–39.79 Mb for H. anomala and 9.92–51.78 Mb for E. splendida (Fig. 1).

Genome-wide chromosomal interactive heatmap. Each chromosome and contig is framed in blue and green, respectively. (a) Himalopsyche anomala. (b) Eubasilissa splendida.
Thorough examination for potential contaminants was conducted using MMseqs. 2 v1125 with the parameter “–min-seq-id 0.8” against the National Center for Biotechnology Information (NCBI) nt and UniVec databases. Sequences with > 90% alignments were removed. The final assembly lengths were 663.43 Mb (H. anomala) and 859.28 Mb (E. splendida), respectively (Table 1). To identify sex chromosomes, Illumina reads of the female individual were mapped against the assembly, and sequencing depth for each chromosome was calculated. Trichoptera follows the ZO female sex determination system26, hence, chromosomes with half the sequencing depth were identified as sex chromosomes (Tables S1, S2). The GC content of H. anomala and E. splendida assemblies was 31.55% and 32.76%, respectively. Notably, the estimated genome size closely matched the assembly size, with the genome assembly size of H. anomala resembling that of other Himalopsyche species27,28, whereas the genome size of E. splendida exceeded that of Eubasilissa regina (440.07 Mb)29. Genome completeness was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.230, employing the parameter “-m genome”, during each stage of the assembly. The completeness was computed as 98.1% and 98.2% for H. anomala and E. splendida, respectively, indicating high-quality assembled genomes (Table 2).
Repetitive sequence and noncoding RNAs annotation
RepeatModeler v2.0.231 and the LTR discovery pipeline (-LTRstruct) of genome tools32 were used to build a de novo repetitive element database. Subsequently, we merged this database with the known repeat element database (Repbase-2018102633 and Dfam 3.134). RepeatMasker v4.0.735 was used to annotate the repeat elements of the two assemblies based on the custom database, identifying 288.10 Mb (approximately 43.43%) and 471.23 Mb (approximately 54.84%) of repetitive sequences for H. anomala and E. splendida, respectively. Among these elements, the largest proportion comprised unclassified elements, accounting for 21.43% and 28.44% of the total genomes of the respective species. Details regarding other common repetitive elements are provided in Tables S3, S4. To annotate the non-coding RNAs, we employed Infernal v1.1.436 and tRNAscan-SE v2.0.937, low-confidence tRNAs by setting parameter “EukHighConfidenceFilter” was filtered. A total of 717 ncRNAs and 766 ncRNAs were annotated in the H. anomala and E. splendida genomes, respectively, with tRNAs constituting more than 50% (384 and 420) of these ncRNAs. Details regarding other noncoding RNAs are provided in Tables S5, S6.
Genome annotation
We integrated a multifaceted approach encompassing ab initio predictions, homologous proteins, and transcriptomic strategies to predict gene structures in the H. anomala and E. splendida genomes. Initially, we used BRAKER v2.1.638, which integrated results from Augustus v3.3.339 and GeneMark v4.3240. In this process, we utilized the arthropod reference proteins from OrthoDB10 v1041 to proceed ab initio predictions. Additionally, we downloaded the protein sequences of model organisms and closely related species (Table 3), including Drosophila melanogaster Meigen, Bombyx mori (Linnaeus), Spodoptera litura (Fabricius) and so on. These sequences were used for homologous gene prediction, employing GeMoMa v1.7.142 with the parameter “GeMoMa.c = 0.5 GeMoMa.p = 10”. Transcriptome sequencing reads underwent the same quality control methods used for DNA sequencing. Subsequently, HISAT2 v2.2.043 and samtools were employed to produce BAM alignments for reference assembly, and StringTie v2.1.644 was used to perform transcriptome assembly. Conclusively, we used MAKER v3.01.0345 to synthesize the three distinct strategies. A total of 11,469 and 10,554 PCGs were predicted in the H. anomala and E. splendida genomes, respectively (Table 4). The average number of exons and introns per gene was similar in H. anomala (9.4 exons and 8.2 introns) and E. splendida (7.1 exons and 8.3 introns). Variations in gene density were observed across different chromosomes, with the highest gene density on chromosome 21 and chromosome 23 in the H. anomala and E. splendida genomes, respectively (Fig. 2a,b). BUSCO was employed to predict protein sequence for both genomes with integrity of 98.4% in protein model, attesting to the high-quality annotation of the genomes.

Characterization of the assembled Himalopsyche anomala and Eubasilissa splendida genome, phylogenetic relationship, and gene family evolution. (a) Himalopsyche anomala. (b) Eubasilissa splendida. From the inner to outer layers: gene density, GC content (GC), DNA transposons (DNA), long-interspersed elements (LINE), long-terminal repeat elements (LTR), short-interspersed elements (SINE), chromosome length (Chr).
To functionally annotate the PCGs, Diamond v2.0.11.14946 was applied to search against the UniProtKB database47, using a sensitive strategy. Furthermore, eggNOGmapper v2.0.148 was used to annotate protein domains based on eggNOG v5.049. Concurrently, InterProScan 5.53–87.050 was also employed to identify domains by Pfam51, SMART52, Superfamily53, Gene3D54, and CDD55 databases. Integration of the predicted results led to the functional annotation of 10,715 (93.42%) and 9,947 (94.24%) PCGs for H. anomala and E. splendida, respectively (Table S7).
Data Records
The newly assembled genomes are available at the NCBI under the BioProject IDs: PRJNA749930 (H. anomala) and PRJNA749861 (E. splendida). Raw Illumina, PacBio, Hi-C, and transcriptome data for both species have been deposited in the Sequence Read Archive under identification numbers SRP351561 (H. anomala)56 and SRP351440 (E. splendida)57. The chromosomal assemblies of H. anomala and E. splendida have been deposited in the NCBI assembly with the accession numbers JAHZMQ00000000058 and JAHZML00000000059, respectively. Results of annotation for repetitive elements and gene prediction for both species are available in the figshare database60.
Technical Validation
We evaluated the quality of H. anomala and E. splendida genome assemblies, focusing on completeness and accuracy. The completeness of assembly was evaluated using BUSCO with the insects_odb10 database, yielding final assemblies with BUSCO completeness of 98.1% and 98.2% for H. anomala and E. splendida, respectively, affirming the high quality of these genomes. To verify accuracy of assembly, we calculated mapping rates by aligning PacBio and Illumina reads to the final assembly: for H. anomala, 96.21%, 96.99%, and 96.41% of reads were successfully mapped, respectively; for E. splendida, higher mapping rates of 96.99%, 97.11%, and 96.42% were obtained, respectively. The Hic assembly underwent manual correction to ensure accuracy, and the Hi-C heatmap showed a well-organized interaction pattern at the chromosomal level (Fig. 1). Additionally, the final annotated gene BUSCO completeness was 98.4% for both H. anomala and E. splendida. Collectively, these results confirm the high quality and accuracy of the new chromosome-level assemblies.
Code availability
No specific code was used in this study. All analytical processes were executed according to the manuals and protocols of the corresponding bioinformatic tools.
References
Dijkstra, K. D., Monaghan, M. T. & Pauls, S. U. Freshwater biodiversity and aquatic insect diversification. Annu. Rev. Entomol. 59, 143–163 (2014).
Morse, J. C. Trichoptera World Checklist. http://entweb.clemson.edu/database/trichopt/index.htm. (2023).
Wiggins, G. B. Caddisflies: the underwater architects. University of Toronto Press. (2004).
Hamid, S. A. & Che, S. Application of aquatic insects (Ephemeroptera, Plecoptera and Trichoptera) in water quality assessment of malaysian headwater. Trop. Life Sci. Res. 28, 143–162 (2017).
Morse, J. C. et al. Freshwater biomonitoring with macroinvertebrates in East Asia. Front Ecol Environ. 5, 33–42 (2007).
Morse, J. C, Frandsen, P. B, Graf, W. & Thomas, J. A. Diversity and ecosystem services of Trichoptera. Diversity and ecosystem Services of Aquatic Insects (ed. by Morse, J. C. & Adler, P. H.). Insects. 10, 125 (2019).
Thomas, J. A., Frandsen, P. B., Prendini, E., Zhou, X. & Holzenthal, R. W. A multigene phylogeny and timeline for Trichoptera (Insecta). Syst. Entomol. 45, 670–686 (2020).
Ge, X. et al. Massive gene rearrangements of mitochondrial genomes and implications for the phylogeny of Trichoptera (Insecta). Syst. Entomol. 48, 278–295 (2023).
Malm, T., Johanson, K. A. & Wahlberg, N. The evolutionary history of Trichoptera (Insecta): A case of successful adaptation to life in freshwater. Syst. Entomol. 38, 459–473 (2013).
Wiggins, G. B. The caddisfly family Phryganeidae (Trichoptera). University of Toronto Press. (1996).
de Moor, F. C. & Ivanov, V. D. Global diversity of caddisflies (Trichoptera: Insecta) in freshwater. Hydrobiologia. 595, 393–407 (2008).
Hjalmarsson, A. E. et al. Molecular phylogeny of Himalopsyche (Trichoptera, Rhyacophilidae). Syst. Entomol. 44, 973–984 (2019).
Jannot, J. E., Bruneau, E. & Wissinger, S. A. Effects of larval energetic resources on life history and adult allocation patterns in a caddisfly (Trichoptera: Phryganeidae). Ecol Entomol. 32, 376–383 (2007).
Luo, S., Tang, M., Frandsen, P. B., Stewart, R. J. & Zhou, X. The genome of an underwater architect, the caddisfly Stenopsyche tienmushanensis Hwang (Insecta: Trichoptera). GigaScience. 7, giy143 (2018).
Ge, X. et al. The First Chromosome-level Genome Assembly of Cheumatopsyche charites Malicky and Chantaramongkol, 1997 (Trichoptera: Hydropsychidae) Reveals How It Responds to Pollution. Genome. Biol. Evol. 1410, evac136 (2022).
Liu, Y. et al. Apolygus lucorum genome provides insights into omnivorousness and mesophyll feeding. Mol. Ecol. Resour. 21, 287–300 (2020).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/. (accessed on 1 October 2022) (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 11, 1432 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Yoshido, A. et al. Step-by-step evolution of neo-sex chromosomes in geographical populations of wild silkmoths, Samia cynthia ssp. Heredity. 106, 614–624 (2011).
Deng, X. L. et al. The impact of sequencing depth and relatedness of the reference genome in population genomic studies: A case study with two caddisfly species (Trichoptera, Rhyacophilidae, Himalopsyche). Ecol. Evol. 12, e9583 (2022).
Heckenhauer, J. et al. Genome size evolution in the diverse insect order Trichoptera. GigaScience. 11, giac011 (2022).
Heckenhauer, J. et al. Characterization of the primary structure of the major silk gene, h-fibroin, across caddisfly (Trichoptera) suborders. iScience. 26, 107253 (2023).
Waterhouse, R. M. et al. BUSCO. Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Gremme, G. The GENOMETOOLS genome analysis system. http://genometools.org. (2023).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA. 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic. Acids. Res. 44, D81–D89 (2016).
Smit, A. F. A, Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (2013–2015) (accessed on 1 October 2022).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods. Mol. Biol. 1962, 1–14 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP and AUGUSTUS supported by a protein database. Nar. Genom. Bioinform. 3, lqaa108 (2021).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic. Acids. Res. 32, W309–W312 (2004).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform. 2, lqaa26 (2020).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic. Acids. Res. 47, D807–D811 (2019).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods. Mol. Biol. 1962, 161–177 (2019).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome. Biol. 20, 278 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC. Bioinform. 12, 491 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Morgat, A. et al. Enzyme annotation in UniProtKB using Rhea. Bioinformatics. 36, 1896–1901 (2020).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by egg NOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic. Acids. Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic. Acids. Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic. Acids. Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic. Acids. Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic. Acids. Res. 45, D200–D203 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP351561 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP351440 (2023).
Ge, X. Y., Peng, L., Sun, C. H. & Wang, B. X. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_031772345.1 (2023).
Ge, X. Y., Peng, L., Sun, C. H. & Wang, B. X. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_031772225.1 (2023).
Ge, X. Y. Chromosome-scale genomes of two caddisflies (Himalopsyche anomala and Eubasilissa splendida). figshare https://doi.org/10.6084/m9.figshare.24305380 (2023).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (32271631; 32370489; 32311520285).
Author information
Authors and Affiliations
Contributions
X.G., C.S. and B.W. conceived and designed the experiments. X.G. and J.D. collected the samples. X.G., L.P. and Z.D. analyzed the data and results. X.G. wrote the manuscript. X.G., C.S. and B.W. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ge, X., Peng, L., Deng, Z. et al. Chromosome-scale genome assemblies of Himalopsyche anomala and Eubasilissa splendida (Insecta: Trichoptera). Sci Data 11, 267 (2024). https://doi.org/10.1038/s41597-024-03097-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03097-3
