
Abstract
Meghimatium bilineatum is a notorious pest land slug used as a medicinal resource to treat ailments in China. Although this no-model species is unique in terms of their ecological security and medicinal value, the genome resource of this slug is lacking to date. Here, we used the Illumina, PacBio, and Hi-C sequencing techniques to construct a chromosomal-level genome of M. bilineatum. With the Hi-C correction, the sequencing data from PacBio system generated a 1.61 Gb assembly with a scaffold N50 of 68.08 Mb, and anchored to 25 chromosomes. The estimated assembly completeness at 91.70% was obtained using BUSCO methods. The repeat sequence content in the assembled genome was 72.51%, which mainly comprises 34.08% long interspersed elements. We further identified 18631 protein-coding genes in the assembled genome. A total of 15569 protein-coding genes were successfully annotated. This genome assembly becomes an important resource for studying the ecological adaptation and potential medicinal molecular basis of M. bilineatum.
Similar content being viewed by others
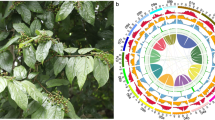

Background & Summary
The Meghimatium bilineatum (syn. Philomycus bilineatus Benson, 1842) is a member of the Philomycidae family and is a notorious quarantine pest land slug that can cause enormous damage to commercial crops, horticultural crops, grasslands, and forests in East Asia1,2,3,4,5. It has a strong ecological adaptation to terrestrial environments and has been widely distributed in various regions of China6. It does not only feed on stems, leaves, fruits, or juices of plants causing direct economic losses but also secretes mucus and excretes feces contaminating fruits and vegetables. This contamination results in a reduction in the market value of products and transmits diseases. Thus, it poses great harm to local agricultural productivity and ecological security, resulting in substantial economic and ecosystem losses7. However, from another perspective, M. bilineatum also exhibits medicinal properties. For example, its crude extracts are used in the treatment of bacterial-induced infectious diseases, the polysaccharides in slug cell are used as natural antioxidants to prevent cancer, and the antimicrobial peptide derived from the slug is utilized to combat skin infections caused by Candida albicans8,9,10. At present, some researchers have carried out in-depth studies on the pharmacological effects of slug extract, indicating that slugs can be used as a valuable medicinal resource with development and application value9,10. Thus, the study of slug species is very meaningful.
In addition to its ecological threat and medicinal value, M. bilineatum, as a member of 30000 described terrestrial gastropod mollusks with shell-less, has completed the transition from aquatic to terrestrial. Similar to other slug species, they have developed many various robust features, including a pulmonate for breathing air, a sophisticated neural-immune system, and the ability to produce mucus to adapt to the terrestrial environments11,12,13. However, compared with land snails, land slugs display unique life strategy for terrestrial environments, such as defense by secreting mucus including specific chemical compounds and better mobility under predation, because they have no protective shell1,14. Furthermore, shell-less land slugs do not expend energy ingesting large amounts of calcium, enabling them to grow faster. Although land slugs have strong adaptation mechanism, their evolutionary history remains unclear. In recent years, molecular phylogenetics analysis of land slugs of the genus Meghimatium based on the mitogenome and nuclear loci has offered new perspectives into the taxonomic revisions and evolution of these species15,16,17. However, these studies cannot fully explain the molecular mechanism of wide ecological adaptation information and the potential genetic basis of medicinal resource traits of this slug. Furthermore, the Philomycidae slug genomics have yet to be published. Therefore, assembling a genome of this slug species should be urgently assembled.
The study of genomes in certain terrestrial mollusks, has shown advancements, including the release of genomic data for two land snails, Achatina fulica and Pomacea canaliculata. However, thorough investigations into the evolutionary mechanisms associated with terrestrial adaptation remain scant18,19. Recently, one genome study of Achatina immaculata, namely giant African snail has verified that some genes related to respiratory system, dormancy system, and immune system have undergone great expansion to adapt to the terrestrial environments20. However, to date, high-quality genomic resources for land slugs are rarely reported. The land slugs and snails, as terrestrial gastropod mollusks with or without shell protection, have different biological processes related to their terrestrial lifestyle. Hence, assembling a genome of the land slug species would facilitate intensive study of this species’ adaptive evolution.
Herein, we assembled the genome of M. bilineatum by uniting the sequencing techniques of Illumina, PacBio, and Hi-C. Three methods, including ab initio gene prediction, homolog and RNA-Seq-based prediction, were used to perform genomic annotation. In addition, the comparative genomics analysis of M. bilineatum and 11 other distantly related species were performed. This study offers insights for the effective management and utilization of slug populations and provides valuable genome information into the evolutionary history and genetic mechanisms of this important gastropod group.
Methods
Land slug collecting and sequencing
Adult land slugs M. bilineatum were collected from a wild area in Zhoushan, Zhejiang, China (122.212 E, 29.979 N). Total DNA was extracted from whole body of the land slug M. bilineatum using the SDS-based extraction method. Then, the DNA samples were purified using QIAGEN® Genomic kit (QIAGEN, Germany) for genome sequencing. First, Illumina short-read library with insert sizes of 300–350 bp was generated, and was sequenced using the Illumina Novaseq. 6000 platform. Second, PacBio HiFi-read library with insert sizes of 10–40 kb was generated using SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA) and sequenced using the PacBio Sequel II platform. Finally, Hi-C short-read library was generated using the purified DNA from the whole body of M. bilineatum according to the previously performed protocol by Belton et al. with given adjustments; it was sequenced using the Illumina Novaseq. 6000 platform21. A total of 250.12 Gb of clean Illumina short-reads, 71.33 Gb HiFi CCS reads and 140.69 Gb clean Hi-C reads were obtained (Table 1).
Total RNA was isolated from whole body of the land slug using TRIzol reagent (Invitrogen, MA, USA) for transcriptome sequencing. The RNA-seq library was generated using NEBNext® Ultra™ RNA Library Prep Kit (NEB, USA) and sequenced using the Illumina Novaseq. 6000 platform. The RNA-seq reads were used for genome annotation. A total of 21.79 Gb of clean data was obtained (Table 1).
Genome size estimation
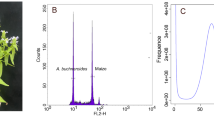
Based on 250.12 Gb clean Illumina short-reads, the genome size, heterozygosity and repetitive sequence content was determined using the k-mer analysis with GCE (1.0.0) following the default parameter22. A total of 223,346,880,670 k-mers with a depth of 144 was obtained (Fig. 1). In addition, the genome size of M. bilineatum was approximately 1.5 Gb, with a heterozygosity of 1.05% and proportion of repeat sequences at 43.69%.

K-mer (17-mer) distribution and estimation of genome size of M. bilineatum.

Chromosomal Hi-C heatmap of the M. bilineatum genome assembly.
Chromosomal-level genome assembly
In the initial genome assembly, HiFiasm (v0.16.0) method was used for ab initio to assemble the genome using the HiFi reads from PacBio23. This preliminary assembly yielded a genome size of 1.80 Gb (Table 2). Subsequently, the redundant sequences were filtered out using Purge_Haplotigs (v1.0.4) software with the parameter of cutoff ‘-a 70 -j 80 -d 200’24. Based on PacBio sequencing data, a 1.63 Gb contig-level genome assembly of M. bilineatum was obtained, and 2526 contigs displayed contig N50 and N90 sizes of 1.37 and 320.449 Mb, respectively (Table 2). The chromosome-level assembly of M. bilineatum was conducted using Hi-C technology. Initially, Bowtie2 (v2.3.4.3) following the default parameters was used to match the 140.69 Gb clean Hi-C reads to the contig-level genome to obtain unique mapped paired-end reads25. A total of 185.36 million paired-end reads were uniquely mapped (Table S1), of which 88.02% represented valid pairs (Table S2). Subsequently, contigs were assembled into the chromosome-level scaffolds using the 3D-DNA processes (v180922) (parameters: -r 0) with all valid pairs, and the JuiceBox (v1.11.08) was used to correct the errors in the genome assembly26,27. We anchored and obtained 25 pseudo-chromosomes with seven unanchored scaffolds. The 25 pseudo-chromosomes covering ~99.95% of the final genome with size ranging from 25.66 Mb to 135.71 Mb (Fig. 2; Table 3). Ultimately, we obtained a 1.61 Gb chromosomal-level genome assembly of M. bilineatum with contig N50 size and scaffold N50 size of 1.36 Mb and 68.08 Mb, respectively. Genome assembly results showed that the genome size of M. bilineatum is similar to that of the Spanish slug Arion vulgaris (1.54 Gb) in the previous study28.
Repeat-content identification and classification
Repetitive sequences, including tandem repeats and interspersed repeats, in M. bilineatum genome were determined using the de novo prediction and homolog-based methods. Based on homology comparison, RepeatMasker (open-4.0.9) (parameters: default) and RepeatProteinMask (parameters: default) software were utilized to find the interspersed repeats against the RepBase database (http://www.girinst.org/repbase)29. On the basis of de novo prediction, TRF (v4.09) software (parameters: default) was used to identify the tandem repeats30. In addition, a repetitive sequence library was constructed using the RepeatModeler (open-1.0.11) with default parameters and LTR-FINDER_parallel (v1.0.7) with default parameters31,32. Then, the RepeatMasker (open-4.0.9) with default parameters was used to identify the repeat element against this repeat library31. After combining the results from de novo prediction and homolog-based methods, we identified and classified 1.18 Gb of repetitive sequences, taking up 72.51% of the assembled genome, mainly including 7.99% DNA elements, 34.08% long interspersed elements (LINE), and 16.35% unknown sequences (Tables 4 & 5). The repeat-content in the M. bilineatum genome is similar to the Spanish slug A. vulgaris (75.09%), and is higher than other studied gastropod species28,33. These results further validate the accuracy of our genome assembly.
Identification and annotation of protein-coding genes
First, we used repeat-masked genome sequences to perform ab initio gene prediction, and then used AUGUSTUS (v3.3.2), Genscan (v1.0) and GlimmerHMM (v3.0.4) software to detect the protein-coding genes34,35,36. Second, to conduct homology-based prediction, protein sequences from Candidula unifasciata (GCA_905116865.2), Elysia chlorotica (GCA_003991915.1), Haliotis rubra (GCA_003918875.1), Haliotis rufescens (GCA_023055435.1), Lottia gigantea (GCA_000327385.1), Pakobranchus ocellatus (GCA_019648995.1), and Pomacea canaliculate (GCA_003073045.1) were compared with the M. bilineatum genome utilizing TBLASTN (v2.2.29) (e-value ≤ 1e-5) to determine candidate regions, and further used GenWise (v2.4.1) software to accurately map the screened proteins to the M. bilineatum genome to obtain splice sites37. Third, to perform transcriptome sequencing-based prediction, the RNA-seq reads from Illumina were mapped to the M. bilineatum genome by using the TopHat (v2.1.1) software following default arguments, and the transcripts were assembled using Cufflinks (v2.2.1) software with the “-e 100 -C” parameter38,39, and the protein-coding genes were determined using the PASA (v2.3.2)40. Fourth, using the MAKER2 (v2.31.10) and HiFAP software following default parameters, we combined the three predictions to construct a complete and nonredundant reference gene database41. Finally, in the M. bilineatum genome, 18631 identified protein-coding genes were found. The length of the average gene, including CDS, exon, and intron, is presented in Table 6. These predicted gene structures were also compared with the seven other homologous species (Fig. 3).

Comparison of protein-coding genes annotation quality. Eight species (M. bilineatum, Haliotis rufescens, Pakobranchus ocellatus, Lottia gigantea, Candidulaunifasciata, Elysia chlorotica, Haliotis rubra, and Pomacea canaliculate) were examined to compare the lengths of the gene, CDS, exon, and intron.
We annotated these protein-coding genes functions through the alignment of gene sequences to the InterPro, GO, KEGG, SwissProt, TrEMBL, TF, Pfam, NR, and KOG database by using BLAST + (2.11.0) software (e-value ≤ 1e-5)42,43,44,45,46,47. In addition, based on InterPro database and Pfam database, the conserved protein domain and motif associated with the function annotated was determined using the InterProScan tool (v5.61-93.0) with the “-seqtype p -formats TSV -goterms -pathways -dp” parameter48. Ultimately, a total of 15569 genes (83.57%) were successfully annotated (Table 7).
Identification of non-coding genes
The tRNA, rRNA, miRNA, and snRNA non-coding RNAs are not translated into proteins. In the annotation process of non-coding RNAs, tRNAscan-SE (v1.3.1) software following the default parameters was used to find the tRNA sequences in the assembled genome according to the structural characteristics of tRNA49. BLASTN was applied to identify rRNA genes in the assembled genome according to the highly conserved characteristics of rRNA. In addition, according to the covariance model of Rfam database (v14.8), we used the INFERNAL program with default arguments to predict the miRNA and snRNA sequences50. Finally, 1424 rRNAs, 941 tRNAs, 588 snRNAs, and 49 miRNAs were annotated (Table 8).
Comparative genomic analysis
The single-copy ortholog genes of M. bilineatum and 11 other molluscan species (Table S3), including Nautilus pompilius, Octopus minor, Bathymodiolus platifrons, Chrysomallon squamiferum, Elysia chlorotica, Biomphalaria glabrata, Candidula unifasciata, Pomacea canaliculate, Haliotis rubra, Gigantopelta aegis and Lottia gigantea, were determined using the “-l 1.5” parameter of hcluster_sq software from OrthoMCL (v2.0.9) to validate the phylogenetic relationships among the 12 molluscan species51. A total of 29157 gene families were determined, including 671 common orthologous gene families and 135 single-copy gene families, in the 12 molluscan species (Fig. 4; Table S4). The MAFFT (v7.487) software with default parameters was used to compare the single-copy genes52. All conserved sequences in the single-copy genes were extracted using Gblock (v0.91b) software with the “-t = c” parameter53. Subsequently, the ML phylogenetic tree was constructed using the “-f a -N 100 -m GTRGAMMA” parameter of RAxML (v8.2.12)54, with N. pompilius and O. minor as the outgroup. Moreover, the divergence time of the 12 mollusks were estimated using the MCMCtree (v4.4) program in software PAML (v4.9) with “clock = 3; model = 0” parameter according to the calibration times of N. pompilius-B. platifrons (619.1–527.6 MYA), B. platifrons-P. canaliculata (541.7–463.4 MYA), N. pompilius-O. minor (452.6–364.2 MYA), B. glabrata-P. canaliculata (496.0–310.0 MYA) and G. aegis-C. squamiferum (100.0–42.4 MYA) from the Timetree database55. The evolutionary tree showed that M. bilineatum and C. unifasciata were clustered together, and diverged ~231.4 MYA (Fig. 5). We also identified the expanded genes and contracted gene families in the 12 mollusks using CAFE (v5.0.0) with the “-p 0.05 -t 4 -r 10000” parameter56. The result showed that there were 879 expanded gene families and 1385 contracted gene families in the M. bilineatum (Fig. 5).

Distribution of genes in different species.

Phylogenetic analysis of M. bilineatum and 11 other mollusks. The green and red numbers on each branch represent the number of significantly expanded and contracted gene families, respectively. The blue numbers on each branch represent the divergence time (MYA) of these 12 mollusks.
Data Records
All sequencing data from three sequencing platforms have been uploaded to the NCBI SRA database (transcriptomic sequencing data: SRR2586702857, genomic Illumina sequencing data: SRR2590398958, genomic PacBio sequencing data: SRR2591904459 and SRR2591904360, Hi-C sequencing data: SRR2591915561 and SRR2591915462). The final chromosome-level assembled genome file has been uploaded to the GenBank database under the accession JAXGFX00000000063. Genome annotation files (including repeat-content annotation, gene structure annotation, gene functional annotation and non-coding genes annotation) have been uploaded to the Figshare database64.
Technical Validation
Evaluating quality of the DNA and RNA
Prior to the genome sequencing, we used the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, San Jose, CA, USA) and Qubit 3.0 Fluorometer (Thermo Fisher Scientific, San Jose, CA, USA) to determine the quality (OD260/280 and OD260/230) and concentration of the DNA and RNA samples to ensure the accuracy of sequencing data. We also used the agarose gel electrophoresis and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, California, USA) to determine the integrity of the DNA and RNA samples.
Evaluating quality of the genome assembly
To evaluate the sequence consistency and assembly quality, the BWA (v0.7.17-r1188) and Minimap2 (v2.24_x64-linux) software were used to map the short reads from Illumina and HiFi reads from PacBio to the assembled genome, respectively65,66. After these processes, 99.35% of the short reads from Illumina and 99.62% of the HiFi reads from PacBio were aligned, covering 99.81% and 99.99% of the assembled genome, respectively (Table S5 & S6). Moreover, BUSCO (v5.4.3) analysis was conducted to evaluate the assembly quality based on the mollusca_odb10 database67. A total of 91.70% of the 5295 single-copy orthologs in the assembled genome were determined as complete, including 4015 single-copy (75.80%) and 842 duplicated (15.90%), 0.89% and 7.46% of the total single-copy orthologs were fragmented and missing, respectively (Table 9).
Evaluating quality of the genome annotation
BUSCO (v5.4.3) analysis was conducted to evaluate the genome annotation quality based on the mollusca_odb10 database67. A total of 91.60% of the 5295 single-copy ortholog genes in the assembled genome were determined as complete, including 3912 single-copy genes (73.90%) and 939 duplicated genes (17.70%), 1.30% and 7.10% of the total genes were fragmented and missing, respectively (Table 9).
Code availability
No specific code was used in this study. The standard bioinformatic tools were used for data analysis. Furthermore, the parameter setting of the bioinformatics tools was performed in accordance with the manual and protocols and described in the Methods Section.
References
Barker, G. The biology of terrestrial molluscs. 1–146 (CABI Wallingford UK, 2001).
Tsai, C.-L. & Wu, S.-K. A new Meghimatium slug (Pulmonata: Philomycidae) from Taiwan. Zool. Stud. 47, 759–766 (2008).
Orians, C. M., Fritz, R. S., Hochwender, C. G., Albrectsen, B. R. & Czesak, M. E. How slug herbivory of juvenile hybrid willows alters chemistry, growth and subsequent susceptibility to diverse plant enemies. Ann. Bot. 112, 757–765 (2013).
Park, G.-M. A new species and a new record of Meghimatium Slugs (Pulmonata: Philomycidae) in Korea. J. Environ. Biol. 39, 399–405 (2021).
Xu, Z. W., Wang, X. F., Wei, X. M. & Shi, H. Ecological observation on Phiolomycus bilineatus and preliminery study on its damage control. Chin. J. Zool 2, 5–8 (1993).
Wiktor, A., De-Niu, C. & Ming, W. Stylommatophoran slugs of China (Gastropoda: Pulmonata)-Prodromus. Folia Malacol 8, 3–35 (2000).
Dong, Y. H., Qian, J. R. & Xu, P. J. Occurrence law of Philomycus bilineatus and its prevention. Acta Agric. Jiangxi 20, 37–38 (2008).
Li, Z., Yuan, Y., Meng, M., Hu, P. & Wang, Y. De novo transcriptome of the whole-body of the gastropod mollusk Philomycus bilineatus, a pest with medical potential in China. J. Appl. Genet. 61, 439–449 (2020).
He, R., Ye, J., Zhao, Y. & Su, W. Partial characterization, antioxidant and antitumor activities of polysaccharides from Philomycus bilineatus. Int. J. Biol. Macromol 65, 573–580 (2014).
Li, Z. et al. In vitro and in vivo activity of phibilin against Candida albicans. Front. Microbiol. 13, 862834 (2022).
Hiong, K. C., Loong, A. M., Chew, S. F. & Ip, Y. K. Increases in urea synthesis and the ornithine–urea cycle capacity in the Giant African Snail, Achatina fulica, during fasting or aestivation, or after the injection with ammonium chloride. J. Exp. Zool. A Comp. Exp. Biol. 303, 1040–1053 (2005).
Mukherjee, S., Sarkar, S., Munshi, C. & Bhattacharya, S. The uniqueness of Achatina fulica in its evolutionary success. in Organismal and Molecular Malacology (ed. Ray, S.) 219–232 (IntechOpen, 2017).
Rosenberg, G. A new critical estimate of named species-level diversity of the recent Mollusca. Am. Malacol. Bull. 32, 308–322 (2014).
Ponder, W. & Lindberg, D. R. Phylogeny and Evolution of the Mollusca. (University of California Press, 2008).
Yang, T. et al. The complete mitochondrial genome sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and phylogenetic analysis. Genes 10, 198 (2019).
Xie, G.-L. et al. A novel gene arrangement among the Stylommatophora by the complete mitochondrial genome of the terrestrial slug Meghimatium bilineatum (Gastropoda, Arionoidea). Mol. Phylogenet. Evol. 135, 177–184 (2019).
Ito, S. et al. Taxonomic insights and evolutionary history in East Asian terrestrial slugs of the genus Meghimatium. Mol. Phylogenet. Evol. 182, 107730 (2023).
Liu, C. et al. The genome of the golden apple snail Pomacea canaliculata provides insight into stress tolerance and invasive adaptation. Gigascience 7, giy101 (2018).
Guo, Y. et al. A chromosomal-level genome assembly for the giant African snail Achatina fulica. Gigascience 8, giz124 (2019).
Liu, C. et al. Giant African snail genomes provide insights into molluscan whole‐genome duplication and aquatic–terrestrial transition. Mol. Ecol. Resour. 21, 478–494 (2021).
Belton, J.-M. et al. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Liu, B. H. et al. Estimation of genomic characteristics by analyzing K-mer frequency in de novo genome projects. Quant. Biol 35, 62–67 (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 1–10 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359 (2012).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3, 99–101 (2016).
Chen, Z., Doğan, Ö., Guiglielmoni, N., Guichard, A. & Schrödl, M. Pulmonate slug evolution is reflected in the de novo genome of Arion vulgaris Moquin-Tandon, 1855. Sci. Rep. 12, 14226 (2022).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 1–3 (2019).
Gomes-dos-Santos, A., Lopes-Lima, M., Castro, L. F. C. & Froufe, E. Molluscan genomics: The road so far and the way forward. Hydrobiologia 847, 1705–1726 (2019).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Birney, E., Clamp, M. & Durbin, R. GeneWise and GenomeWise. Genome Res 14, 988–995 (2004).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, 1–13 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666 (2003).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 1–14 (2011).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32, W20–W25 (2004).
Apweiler, R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res 32, D115–D119 (2004).
Finn, R. D. et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res 45, D190–D199 (2017).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199–D205 (2014).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 1–14 (2003).
Bairoch, A. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–D124 (2005).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003).
Nakamura, T., Yamada, K. D., Tomii, K. & Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492 (2018).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25867028 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25903989 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25919044 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25919043 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25919155 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25919154 (2023).
Sun, S. L., Han, X. L., Han, Z. Q. & Liu, Q. Meghimatium bilineatum, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAXGFX000000000 (2023).
Sun, S. L. Chromosomal-scale genome assembly and annotation of the land slug (Meghimatium bilineatum). figshare https://doi.org/10.6084/m9.figshare.24038871.v1 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (LR21D060003) and the Introduction of Talent Research Start-up Fund of Zhejiang Ocean University (JX6311031923).
Author information
Authors and Affiliations
Contributions
Z.Q.H. designed the project. S.L.S., X.L.H. and Q.L. collected the samples and analyzed the data. S.L.S. and Z.Q.H. wrote the manuscript. S.L.S., Z.Q.H. and Q.L. revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, S., Han, X., Han, Z. et al. Chromosomal-scale genome assembly and annotation of the land slug (Meghimatium bilineatum). Sci Data 11, 35 (2024). https://doi.org/10.1038/s41597-023-02893-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02893-7
