
Abstract
Sciaenops ocellatus is among the most important artificially introduced farmed fish across 11 countries and regions. However, the frequent occurrence of extreme weather events and breeding escapes have placed great pressure on local marine biodiversity and ecosystems. We reported the de novo assembly and annotation with a contig N50 of 28.30 Mb using PacBio HiFi sequencing and Hi-C technologies, which resulted in a 283-fold increase in contig N50 length and improvement in continuity and quality in complex repetitive region for S. ocellatus compared to the previous version. In total, 257.36 Mb of repetitive sequences accounted for 35.48% of the genome, and 22,845 protein-coding genes associated with a BUSCO value of 98.32%, were identified by genome annotation. Moreover, 54 hub genes rapidly responding to hypoosmotic stress were identified by WGCNA. The high-quality chromosome-scale S. ocellatus genome and candidate resistance-related gene sets will not only provide a genomic basis for genetic improvement via molecular breeding, but will also lay an important foundation for investigating the molecular regulation of rapid responses to stress.
Similar content being viewed by others
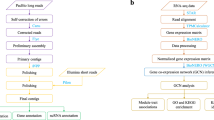

Background & Summary
Red drum (Sciaenops ocellatus, Linnaeus, 1766, FishBase ID: 191), an estuarine fish species native to the western Atlantic Ocean from Massachusetts to northern Mexico in the United States, is one of the most important farmed fish species in the world1,2,3,4,5 (Figure S1). S. ocellatus is a temperate and saline fish with obvious characteristics such as a fast growth rate (≥10 °C, ~26 kg with a length of 1.23 metres according to wild angling in China), miscellaneous eating habits, a strong reproductive ability, high disease resistance, a high survival rate (2–33 °C), and low oxygen tolerance (≥2.2 mg/l), and can adapt to diverse habitats (the bay, intertidal, saline environments, and inland freshwater areas)6,7,8,9. The main breeding areas of S. ocellatus have expanded to 11 countries and regions covering the Atlantic, Pacific, Indian Ocean and Mediterranean regions through the globalization of breeding for more than 30 years, and the total breeding production (as of 2019) has reached 77008.58 tons with a product value of 1.96 × 108 USD (www.fao.org/faostat) (Figure S1). The aquaculture of S. ocellatus has diversified modes mainly including seawater cage aquaculture, factory indoor aquaculture and nearshore pond aquaculture, of which seawater cage aquaculture accounts for more than 90% of the total aquaculture production9,10,11,12. In recent years, it was found that millions of S. ocellatus have escaped from seawater cages in the coastal areas of Zhejiang and Fujian, China, and individuals with mature gonads have been caught in natural marine areas9,12. Meanwhile, there have been increasing reports of S. ocellatus being caught in nonnative waters (off the coast of Mexico), with records in the Indo-West Pacific (e.g., Singapore and Thailand) and the Western Pacific (e.g., Taiwan Strait of China, East China Sea, South China Sea, and Korea)13,14,15,16,17. Wang et al.17 used an environmental DNA approach to detect a high abundance of S. ocellatus in the Jiaojiang Estuary and Sanmen Bay area in the East China Sea. Fishery resource surveys further revealed that alien S. ocellatus in the coastal waters of western Taiwan and the Indo-West Pacific have established breeding populations with significant invasive properites9,15,16. Therefore, it is necessary to obtain a high-quality chromosome-scale assembled genome and abundant resistance-related gene resources, which will facilitate studies of the molecular mechanisms of resistance in S. ocellatus and provide basic resources for assessment the subsequent rapid environmental adaptation and invasion genetics assessment of this species.
It is well known that high-quality reference genomes and complete annotations can provide important tools for population genomics and environmental adaptation genetics studies to efficiently mine genetic resources and accelerate the assessment of environmental adaptation18,19,20,21,22. Currently, only one version of the genome assembly for S. ocellatus has been published on the National Center for Biotechnology Information (NCBI) platform23. However, this version of the S. ocellatus genome was sequenced using the Illumina HiSeq. 2000 approach, which resulted in a heavily fragmented genome with a contig N50 length of only 99.71 kb. Therefore, an improved, high-quality version of the S. ocellatus genome is urgently required to support subsequent studies on the precise exploitation of genetic resources and genetic evaluation. PacBio SMRT sequencing employs a cyclic consensus long-line sequencing strategy to generate highly accurate HiFi reads, which can be combined with Hi-C assembly technology to enhance the continuity and quality of sequences, especially in complex repetitive regions of the S. ocellatus genome. This powerful combination of technologies has the potential to produce a more complete and accurate assembly of the S. ocellatus genome.
The integration of RNA sequencing technology and weighted gene coexpression network analysis (WGCNA) method enables a deeper understanding of the complex molecular mechanisms underlying various biological processes, including development, diseases, and environmental adaptation20,24,25,26,27. The combined analysis of RNA-seq and WGCNA has been successfully applied in the investigation of immune response, reproductive development, growth regulation, stress response mechanisms, and key gene identification in various fish species, such as Siniperca chuatsi26, Sebastes schlegelii25, Cynoglossus semilaevis24, Lateolabrax maculatus27, and Paralichthys olivaceus20. Some progress has been made in understanding the genetic and molecular regulatory mechanisms of the stress response of S. ocellatus under osmotic stress, such as high salt stress, and low salt stress. Although significant changes in expression associated with ion transport-related proteins upon exposure to osmotic stress have been detected for classic genes (e.g. nka, and nkcc), a more comprehensive analysis of gene modularity and connectivity between genes by gene coexpression network analysis has not been performed5,28,29. In this study, key modules and core genes were identified by analysing the relationship between S. ocellatus gene expression modules and low osmotic adaptation traits (32 psu, 16 psu, and 3 psu) through WGCNA, providing more effective data and a direction for supplementing and exploring the adaptation mechanism of S. ocellatus in adverse environments.
In this study, we used a combination of PacBio HiFi long-read sequencing and Hi-C technologies to assemble a high-quality chromosome-level S. ocellatus genome with a contig N50 size of 28.30 Mb. With this version of the high-quality genome assembly, we further improved the annotation of S. ocellatus repetitive sequences and protein-coding genes. In addition, based on the relevant RNA-seq datasets obtained from salinity stress gradient stress experiments, five key modules and 592 candidate genes responsive to hypoosmotic stress were identified by gene coexpression network analysis. The high-quality chromosome-scale assembly of the S. ocellatus genome and the identification of candidate stress-related gene sets not only provide a genomic basis for the genetic improvement of S. ocellatus via molecular breeding, but also lay an important foundation for studying the molecular regulation of the rapid stress response and the mechanism underlying rapid global environmental adaptation in S. ocellatus.
Methods
Sample collection, library construction and sequencing
High-quality genomic DNA was extracted from fresh muscle tissue samples of a single female S. ocellatus from offshore Qingdao, Shandong Province, China (Fig. 1). The extracted DNA concentrations were assayed using a combination of Nanodrop (Thermo, NANODROP2000) and Qubit (Invitrogen, QubitTM3Flurometer) methods, and then DNA integrity was examined using 1.5% agarose gel electrophoresis.

The image represented the adult S. ocellatus used for the genome sequencing and assembly process.
A short library (300–350 bp) was constructed using the NR604-VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina (Vazyme, Illumina TruSeq DNA Library Prep Kit) and paired-end 150 bp (PE 150) sequenced using the standard protocol provided by the Illumina NovaSeq. 6000 platform (Illumina Inc., San Diego, CA, USA), yielding a total of 88.22 Gb of clear reads (90.7 Gb of raw reads) with a Q20 of 96.78% for assessing S. ocellatus genome size (Fig. 2, Table 1). High-molecular weight (HMW) gDNA was prepared into 15-kb libraries for PacBio HiFi read production using a standard protocol provided by PacBio (Pacific Biosciences, USA) (Fig. 2). The genomic libraries were sequenced on two cells using the self-testing high-precision CCS mode available as part of the PacBio Sequel II system. We obtained 54.78 Gb of HiFi long-reads with a read N50 length of 12.40 kb, resulting in 84-fold coverage of the S. ocellatus genome size (Fig. 2, Table 1). Hi-C libraries were generated using a process involving HindIII digestion of cross-linked high-quality DNA, 5′ biotin labelling and flat end repair to form chimeric junctions which were further physically sheared to a size into 300–700 bp fragments30,31. A total of 91.78 Gb (~135.31X) of paired-end clean reads were generated from the Hi-C library and sequenced using 150-bp paired-end sequencing on the Illumina NovaSeq. 6000 platform (Fig. 2, Table 1). Assisted genome annotation of tissue (muscle, gill, kidney, gonad, and liver) RNA Illumina libraries and mixed-tissue RNA CCS sequencing libraries yielded 39.73 Gb and 1.85 Gb of clean reads for genome annotation, respectively (Fig. 2, Table 1).

This diagram illustrates the workflow employed for the genome assembly and annotation of S. ocellatus in this study.
S. ocellatus adults were collected from the aquaculture nets of Fuqiang Aquaculture Company (Shandong Province), China. Thirty-six individuals (1,025 ± 62 g) were transferred to the aquarium of the Institute of Oceanography, Chinese Academy of Sciences for temporary culture for 48 h. These fishes were allocated into nine tanks (400 L, 3 experimental groups × 3 replications) with water temperature, salinity, and light-dark cycle for 22 °C, 32 psu, and 13:11, respectively. After 48 h of temporary culture with aeration and satiety feeding, the fish were gradually exposed to three test experimental salinities (32, 16 and 3 psu) under 22 °C water, where 32 psu was set as the experimental control group. To elucidate the rapid response and in vivo regulation mechanisms in S. ocellatus under acute hypoosmotic stress, we sampled all control and experimental fish exposed to the set salinity in tanks and completed the experiment within 24 h. The fish were euthanized by complete immersion in an MS-222 bath (130 mg/L) followed by transection of the spine. Kidney and lamellae were removed from both branchial arches, which were immediately placed in 2 ml cryopreservation tubes and stored in liquid nitrogen for Illumina sequencing-based transcriptome experiments analysis. Gill and kidney tissues of S. ocellatus obtained from the three gradients of low-salt osmotic stress experiments (32 psu, 16 psu, and 3 psu) were subjected to library construction (300 ~ 500 bp) and sequencing (PE150) using standard protocols provided by the Illumina NovaSeq. 6000 platform. We obtained approximately 138.36 Gb of raw data including 916,304,804 reads for subsequent WGCNA (Fig. 2, Table 2).
Genome survey and assembly
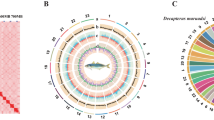
A total of 88.22 Gb of Illumina clear reads was used to estimate the primary characteristics of the genome in SOAPec (version 2.01)32 and GenomeScope (version 2.0)33 software with 19 K-mer frequencies (Figure S2). The survey results showed that the genome size of S. ocellatus was 678.28 Mb with a repetitive sequence ratio, heterozygosity and GC content of 20.54%, 0.45% and 41.11%, respectively (Figure S3). Then, 54.78 Gb of HiFi long reads were used by the Hifiasm (v0.15.1)34 software with default parameters for assembly, and we obtained a genome size of 725.33 Mb comprising 316 contigs with a contig N50 length of 28.30 Mb for S. ocellatus (Table 3). The genome assembly was corrected by LACHESIS using 62.3% of uniquely mapped paired read pairs (259,072,033 pairs) from the Hi-C libraries, with scaffolds placed on chromosomes by clustering, sorting and orientation35,36,37 (Fig. 3). The highly contiguous genomes (contigs N50 26.29 Mb, scaffold N50 33.32 Mb) with a size of 701.63 Mb, accounting for 96.73% of the draft S. ocellatus genome, was assembled into the 24 corresponding chromosomes based on the karyotype analyses, which demonstrated the higher quality of this genome assembly compared to other previously published versions for further studying and understanding S. ocellatus species (Fig. 3, Table 3, Table 4).

The heatmap displays the genome-wide Hi-C interactions in the assembly of the S. ocellatus genome.
The quality of the contig version of the S. ocellatus genome assembly was evaluated using CEGMA (v2.5)38 and BUSCO (v4.0, actinopterygii_odb10)39 assessments. The CEGMA assessment showed that 99.56% of the Core Eukaryotic Genes (CEGs) were present in the genome assembly. The BUSCO assessment indicated that 98.32% of the complete BUSCOs (Benchmarking Universal Single-Copy Orthologs) were present in the assembly (Table 5). TBtools software (v1.131, min_repeat_length 80 bp, max_repeat_length 350 bp, window_size 5000 bp, step_size 1000 bp) was used to confirm that the assembled S. ocellatus genome was composed of telomeric chromosomes, which were consistent with the previously reported chromosome C-banding results for S. ocellatus40,41,42 (Figure S4). Further statistics of genome-wide sequence difference covariance between the present genome version of S. ocellatus and the reported version using TBtools software (v1.131) showed that the assembled S. ocellatus genome was significantly superior to the previous version, displaying 202,685 deletion sites and 191,257 insertion sites42 (Fig. 4).

The covariance of sequence differences between the assembled PacBio genomes of S. ocellatus and the previously reported Illuminar version was shown on a genome-wide scale. (a) The reported S. ocellatus genome version vs the assembled version in the present study. (b) The assembled genome version of S. ocellatus vs the reported version. (c) Distribution of differential sites (assembled genome version of the present study as reference) on chromosomes with (1) Chromosome name, (2) Distribution of the number with deletions at the locus from 0 to 5000 bp, (3) Distribution of the number with deletions at the locus more than 5000 bp, (4) Distribution of the number with insertions at the locus from 0 to 5000 bp, (5) Distribution of the number with insertions at the locus more than 5000 bp.
Genomic repeat annotation
Transposable elements (TEs) were identified by a combination of homology-based and de novo approaches (Fig. 2). The RepeatModeler2 (v2.0.1)43 tool incorporating the RECON (v 1.08)44 and RepeatScout (v1.0.6)45 programs was first used to generate a customized de novo repeat library for the S. ocellatus genome. Then full-length long terminal repeat retrotransposons (fl-LTR-RTs) were identified using both LTRharvest (v1.5.9)46 (-minlenltr 100 -maxlenltr 40000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes) and LTR_finder (v1.1)47 (-D 40000 -d 100 -L 9000 -l 50 -p 20 -C -M 0.9). The high-quality intact fl-LTR-RTs and nonredundant LTR library were then produced by LTR_retriever (v2.8)48. A nonredundant species-specific TE library was constructed by combining the de novo TE sequence library with the known Repbase (v19.06)49, REXdb (v3.0)50 and Dfam (v3.2)51 databases. Finally, RepeatMasker (v4.10)52 was employed to perform homology searches of nonredundant TE libraries to identify and classify TEs. Approximately 211,569,554 bp of TE sequences (375 full-length LTR sequences) accounting for 29.17% of the S. ocellatus genome were obtained. There represented an increase of 90.69 Mb compared to the previously reported version (Table 6)23. A total of 45.79 Mb of tandem repeats (6.31% of the whole genome) were also annotated by the Tandem Repeat Sequence Finder (v409, 1 1 2 80 5 200 2000 –d -h)53 and MIcroSAtellite Identification Tool (MISA v2.1)54 (Table 6).
Protein-coding gene identification and functional annotation
For noncoding RNA (ncRNA) annotation, tRNAs and rRNA were identified using tRNAscan-SE (v1.3.1)55 and barrnap (v0.9)56 based on the Rfam (v12.0)57 database, respectively. miRNA was identified using the miRBase database58. snoRNA and snRNA were predicted using Infernal (v1.1)59 based on the Rfam (v12.0)57 database. In total, 2,405 tRNAs, 2,986 rRNAs, and 582 miRNAs were predicted (Fig. 2, Table 7).
We used three approaches for gene prediction in the repeat-masked genome, including ab initio prediction, homology-based search, and transcriptome-based assembly (Fig. 2). Augustus (v2.4)60 and SNAP (2006-07-28)61 software were integrated for ab initio gene prediction and yielded 40,194 and 65,619 genes, respectively (Table 8). For the homologue-based approach, GeMoMa (v1.7)62 software was used with a reference gene model from the Gasterosteus aculeatus63, Gadus morhua64, Larimichthys crocea65, Oryzias latipes66, and Paralichthys olivaceus67 species, which annotated between 8,322 and 19,274 homologous genes (Table 8). Two strategies were applied for transcriptome-based gene annotation, which were comparison followed by assembly and assembly followed by comparison. We integrated Hisat (v2.0.4)68, StringTie (v1.2.3)69, and GeneMarkS-T (v5.1)70 software to map RNA sequence data to a reference genome and generate the assembly to predict 20,175 genes. PASA (v2.0.2)71 software was utilized to predict 20,510 genes using unigenes and full-length transcripts obtained by PacBio sequencing assembled with Trinity (v2.11)72. The gene models from these three approaches were merged using EVM software (v1.1.1)73 and updated by PASA, yielding a final total of 22,845 protein-coding genes with an average of 10.8 exons per gene in the S. ocellatus genome (Fig. 5, Table S1). The Venn diagram further showed that 19,629 genes (85.92%) were derived from transcriptome and homology-based predictions, indicating high quality of gene prediction for S. ocellatus (Figure S5). The BUSCO (v4.0)39 assessment showed that 97.9% of the complete BUSCOs were present in the assembly. This value indicated that the integrity of the assembled genome was high and significantly improved compared to the previously reported version (93.2%, complete BUSCOs) (Table 9). Meanwhile, the present version of the S. ocellatus genome (22,845) included 2,792 more annotated protein-coding genes than the previous version (20,053) (Table 10).

Comparison of genomic elements in closely related species. (a) Coding length distribution; (b) Gene length; (c) Intron length of each gene; (d) Exon length.
The protein-coding genes were functionally annotated by aligning the obtained protein-coding genes with the NCBI Nonredundant protein (NR) (202009, ftp://ftp.ncbi.nlm.nih.gov/blast/db), GO (20200615, http://geneontology.org)74, KEGG (20191220, http://www.genome.jp/kegg)75, SWISS-PROT (202005, http://ftp.ebi.ac.uk/pub/databases/swissprot)76, and Pfam (v33.1, http://pfam.xfam.org)77 databases using Blastp (v2.2.26)78 and Diamond (v0.8.22)79 software with an e-value threshold of 1e-5, which yielded the percentage of genes with functional annotations ranging from 69.92% to 99.83% (Table 10). Then, according to the above annotation results combined with the EggNOG (v5.0, http://eggnog5.embl.de/download/eggnog_5.0/) database for further verification and additional annotation of protein-coding genes, a total of 22,812 protein coding genes were functionally annotated (99.86% of the total number of predicted genes) (Table 10). A total of 13,712 of these annotated protein-coding genes received common functional annotations in the eight functional datasets described above (Fig. 6). Protein structural domains and motifs were also annotated using the InterProScan (v5.34-73.0)80 database, yielding 59,941 structural domains and 2,077 motifs, respectively.

Upset plot showing the functional annotation of genes in the assembled genomes of S. ocellatus based on different databases.
Weighted gene coexpression network (WGCA) construction
For high-throughput transcriptome sequencing to obtain raw data, we performed the following standard processing protocol. First, we utilized fastp (v0.18.0)81 and Bowtie2 (v 2.2.8)82 software to filter low-quality and ribosomal RNA (rRNA) data from raw data (138.36 Gb, 916,304,804 reads), respectively. The paired-end clean data (137.25 Gb, 908,288,508 reads) (Q30 ≥ 95.2%) were aligned to the S. ocellatus genome using the Hisat2 (v2.0.5)68,83 software with default parameters, achieving an alignment rate of 95.19% (864,561,166 reads). Then, StringTie (v1.3.1)69 program was employed to determine the count of reads aligned to each gene in the reference genome. Subsequently, fragment per kilobase of transcript per million mapped reads (FPKM) values were calculated using RSEM84 software to estimate gene expression levels. Differential expression analysis between two groups was conducted using the DESeq. 2 R package85 (Fig. 2). According to the expression criterion (|log2fold change| ≥ 1) and a p-adj value < 0.05, we detected a total of 334, 59, 329, 527, 30, and 334 differentially expressed genes (DEGs) in the comparisons of FWG3 vs. CKG32, MSG16 vs. CKG32, FWG3 vs. MSG16, FWK vs. CKK, MSK vs. CKK, and FWK vs. MSK, respectively (Table 2) (Supplementary DEGs_Set.xlsx).
To identify the components of gene modules in response to hypo-osmotic stress for S. ocellatus, we constructed correlations between gene expression modules and hypo-osmotic acclimation traits (intake status, swimming speed and respiratory rate) using the WGCNA method based on the RNA-seq dataset obtained from the salinity stress gradient experiment (32 psu, 16 psu, and 3 psu). The WGCNA (v1.47)86 package in R was used to construct a coexpression network. After filtering out 7,946 genes, 16,034 gene expression values were utilized in WGCNA to create coexpression modules (Figure S6). The automatic network construction function blockwiseModules (power = 7, TOMType = unsigned, mergeCutHeight = 0.1, minModuleSize = 50) was employed with the default settings for the remaining parameters. This resulted in the clustering of genes into 9 related modules, where genes within the same module were considered to exhibit similar expression patterns (Figure S7). We used Pearson’s method to further statistically compute the correlation coefficients for module-to-module correlations and gene-to-module eigenvalue correlations, and utilized Student’s t test to obtain p values. Four significantly correlated module pairs were detected for turquoise-blue (r = 0.67, p < 0.001), red-blue (r = 0.88, p < 0.001), green-yellow (r = 0.65, p < 0.005), and yellow-red (r = 0.64, p < 0.005) (Fig. 7). Five modules significantly correlated with traits (positive correlation: pink module r = 0.47 ~ 0.50; negative correlation: blue module r = −0.55 to −0.57; red module r = −0.58 ~ −0.61; green module r = −0.58 ~ −0.59, and yellow module r = −0.76 ~ −0.8) were identified based on Spearman correlation coefficients between module eigenvalues and traits (Fig. 7).

WGCNA analysis of hypoosmotic stress experiments using RNA high-throughput sequencing. (a) It represented module-to-module relationships, with stronger colours representing significant correlations. (b) Constructed gene co-expression clustering tree and co-expression topology heat map. Note, stronger colours represented higher expression similarity between genes. (c) It referred to the expression pattern samples, which represented the expression trend of the module eigengenes in different samples, and the similarity of the colours represented the consistent expression trend of the sample genes. (d) The constructed relationship matrix of modules-traits, where positive values represented module eigengenes positively correlated with the trait and negative values represented their negative correlation with the trait. Note, significance was taken at 0.05.
Module functional enrichment and identification of key hub genes
To further identify the core module (pink, blue, red, green, and yellow) genes closely associated with hypoosmotic stress traits, we integrated the gene connectivity results within the module (All.kWithin) and used the CentiScape87 plugin in Cytoscape (v3.10.0)88 for core gene selection using the closeness, degree, and betweenness methods. We obtained 41, 168, 105, 150 and 128 core genes for the pink, blue, red, green and yellow modules, respectively, and annotated the biological processes and KEGG pathways of candidate genes in Metascape89 (min_overlap 1, p-value cutoff 0.05, min_enrichment 1.5) (https://metascape.org/) (Supplementary hub_gene_set.xlsx, Figure S8).
GO biological process enrichment of the core genes in the pink module with a positive module–trait correlation indicated that S. ocellatus rapidly initiated protein translation processes, including cytoplasmic translation, biosynthesis of the ribonucleoprotein complex, and the haemoglobin biosynthetic process in response to hypoosmotic environmental stress (Figure S8). KEGG functional enrichment analyses further showed that hypotonic stress induced ribosomal complex assembly and protein translation processes.
GO biological process enrichment results of the core genes in the blue module with a negative module–trait correlation indicated that hypotonic stress initiated biological processes such as cellular response to stress, aerobic respiration, stress granule assembly, apoptotic signaling pathway, and the acetyl-CoA biosynthetic process from pyruvate, in addition to initiating the protein translation process. In addition, KEGG enrichment analysis also confirmed that hypotonic induction initiated pathways such as NOD-like receptor signalling pathways, oxidative phosphorylation, and protein processing in the endoplasmic reticulum (Figure S8).
GO enrichment analysis of the red module also showed that hypotonic stress promoted biological processes such as the ncRNA metabolic process, the biosynthetic process of nucleobase-containing compounds, negative regulation of the MAPK cascade, regulation of cellular response to stress, and response to corticotropin-releasing hormone, in addition to inducing protein translation processes (Figure S9). The KEGG enrichment results further showed that hypotonic stress induced the MAPK signalling pathway, as well as energy metabolism (such as the citrate cycle-TCA cycle) and stress response (protein processing in the endoplasmic reticulum), causing S. ocellatus to have a hypotonic response with a low intake rate, and low respiration rate and so on.
The GO biological process enrichment results for the green module showed that hypotonic stress initiated the cellular mitochondrial structure and electron respiratory chain complex assembly process while promoting the protein translation process, which might be compatible with a decline in salt ion efflux function in vivo and reduction in endogenous energy demand under hypotonic conditions. The KEGG enrichment results confirmed that hypotonicity promotes the citric acid cycle and amino acid metabolism reactions in addition to inducing organismal ribosome assembly as well as oxidative phosphorylation reactions (Figure S10).
Both the GO biological process enrichment and KEGG enrichment results for the yellow module, which was significantly correlated with the red and green modules, confirmed that hypotonic induction promoted ribosome small- and large-subunit biogenesis as well as protein translation, and accelerated DNA-guided transcription and nucleocytoplasmic translocation biological processes (Figure S11).
We intersected the core set of genes in the modules closely associated with the traits obtained in the above analyses with the set of DEGs obtained from the hypotonic induction experiments, and finally obtained a set of 54 key hub genes associated with hypotonic stress tolerance in S. ocellatus. GO enrichment of core DEGs revealed that hypotonic stress promoted cell-autonomous motility and environmental signalling processes, inducing multiplexed metabolic processes (such as the glyoxylate metabolic process, small molecule biosynthetic process, and ribonucleoside monophosphate metabolic process) (Fig. 8). The KEGG enrichment results further revealed that hypotonic stress stimulated the glycine, serine and methionine metabolic antiaging pathways90, as well as the endogenous taurine-synthesized cysteine and methionine metabolic pathways90,91, which in turn contributed to the stress tolerance and environmental adaptation of S. ocellatus.

The biological processes GO enrichment and KEGG enrichment of differentially expressed hub genes. (a) GO enrichment for biological processes of 54 hub genes. (b) KEGG enrichment of 54 hub genes.
Data Records
The sequencing datasets and genome assembly have been deposited into the public database. Genome sequencing data (Illumina, PacBio, Hi-C and RNA-seq data) and transcriptomic sequencing data used for Genome assembly has been deposited in the SRA at NCBI SRP46511192.
The transcriptomic sequencing data for WGCA has been deposited in the SRA at NCBI SRP46508093.
The final chromosome assembly and genome annotation files and associated supplementary results have been deposited in the GCA at Genbank GCA_033000465.194 and Figshare95.
Technical Validation
The Merqury (v1.3)96 software was used to assess the quality of the genome, and the consensus quality value (QV) and completeness statistic values with 53.38 and 93.23% indicated that the assembled genome possessed a high level of accuracy and completeness (Figure S12). The Illumina sequencing reads were also aligned to the assembled genome using BWA (v0.7.8)97. The results showed that 99.44% of the reads were successfully mapped to the assembly, and 98.28% of the reads were properly mapped, indicating a high mapping efficiency (Table 5). The RNA-seq clean data was further aligned to the assembled genome using Hisat2 (v2.0.5)68. The results showed that 83.03% of the transcriptome data mapped to the predicted exons, demonstrating the high accuracy of the prediction model used (Table 9).
Code availability
All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatics software.
References
Ackerly, K.L., Roark, K.J. & Nielsen, K.M. Short-term Salinity Stress During Early Development Impacts the Growth and Survival of Red Drum (Sciaenops ocellatus). Estuaries and Coasts (2022).
Bacheler, N. M., Buckel, J. A. & Paramore, L. M. Density-dependent habitat use and growth of an estuarine fish. Canadian Journal of Fisheries and Aquatic Sciences 69, 1734–1747 (2012).
Forsberg, J.A., Dorsett, P.W. & Neill, W.H. Survival and Growth of Red Drum Sciaenops ocellatus in Saline Groundwaters of West Texas, USA. 27, 462-474 (1996).
Rooker, J. R., Stunz, G. W., Holt, S. A. & Minello, T. J. Population connectivity of red drum in the northern Gulf of Mexico. Marine Ecology Progress Series 407, 187–196 (2010).
Watson, C. J., Nordi, W. M. & Esbaugh, A. J. Osmoregulation and branchial plasticity after acute freshwater transfer in red drum, Sciaenops ocellatus. Comp Biochem Physiol A Mol Integr Physiol 178, 82–9 (2014).
Craig, S. R., Neill, W. H. & Gatlin, D. M. Effects of dietary lipid and environmental salinity on growth, body composition, and cold tolerance of juvenile red drum (Sciaenops ocellatus). Fish Physiology and Biochemistry 14, 49–61 (1995).
Jin, H. W., Xu, H. X., Xu, W. J. & Xue, L. J. Study on feeding habits Sciaenops ocellatus of the escaped red drum in Zhejiang sea area. Marine Fisheries Research 29, 103–108 (2008).
Thresher, R. E. & Kuris, A. M. Options for Managing Invasive Marine Species. Biological Invasions 6, 295–300 (2004).
Xue, L.J. Master, Ocean University of China (2008).
Kang, B. et al. Introduction of non‐native fish for aquaculture in China: A systematic review. Reviews in Aquaculture, 1–28 (2022).
Liu, J. H., Mao, X. H., Wang, W. X. & Wang, B. A preliminary study on technique for fully artificial breeding of red drum Sciaenops ocellatus. Journal of Fishery Sciences of China 5, 4 (1998).
Zhang, B., Ma, N.F. & JIang, Z.Q. Pond Cultural Technique of Sciaenops ocellatus. 21, 2 (2002).
Alidoost Salimi, P. et al. A review of the diversity and impact of invasive non-native species in tropical marine ecosystems. Marine Biodiversity Records 14, 11 (2021).
Jaafar, Z., Yeo, D.C.J., Tan, H.H. & O’Riordan, R.M. Status of estuarine and marine non-indigenous species in Singapore. The Raffles Bulletin of Zoology, 13 (2012).
Liao, Y. C., Chen, L. S. & Shao, K. T. The predatory Atlantic red drum, Sciaenops ocellatus, has invaded the western Taiwanese coast in the Indo-West Pacific. Biological Invasions 12, 1961–1965 (2010).
Lin, B. A. et al. First records of small juveniles of the red drum Sciaenops ocellatus (Linnaeus, 1766) in a subtropical mangrove habitat of China. BioInvasions Records 9, 96–102 (2020).
Wang, X., Zhang, H., Lu, G. & Gao, T. Detection of an invasive species through an environmental DNA approach: The example of the red drum Sciaenops ocellatus in the East China Sea. Sci Total Environ 815, 152865 (2022).
Sun, L., Jiang, C., Su, F., Cui, W. & Yang, H. Chromosome-level genome assembly of the sea cucumber Apostichopus japonicus. Sci Data 10, 454 (2023).
Wang, Y., Zhang, H., Xian, W. & Iwasaki, W. Chromosome genome assembly and annotation of the spiny red gurnard (Chelidonichthys spinosus). Sci Data 10, 443 (2023).
Xu, X. W., Zheng, W., Yang, Y., Hou, J. & Chen, S. High-quality Japanese flounder genome aids in identifying stress-related genes using gene coexpression network. Sci Data 9, 705 (2022).
Yang, T. et al. Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics. Nat Genet 54, 1553–1563 (2022).
Zhou, Y. et al. Chromosome genome assembly and annotation of the yellowbelly pufferfish with PacBio and Hi-C sequencing data. Sci Data 6, 267 (2019).
Xu, T., Li, Y., Chu, Q. & Zheng, W. A chromosome-level genome assembly of the red drum, Sciaenops ocellatus. Aquaculture and Fisheries 6, 178–185 (2021).
Wang, N., Liu, Y., Wang, R. & Chen, S. The linkage of cell cycle and DNA replication with growth difference in female Chinese tongue sole (Cynoglossus semilaevis): Analysis from transcriptomic study and WGCNA. Comp Biochem Physiol Part D Genomics Proteomics 39, 100833 (2021).
Wang, X. et al. Transcriptome Analysis Provides Insights into Copulation, Fertilization, and Gestation in Sebastes schlegelii. Genes (Basel) 13 (2022).
Zhou, Y. et al. The dynamic immune responses of Mandarin fish (Siniperca chuatsi) to ISKNV in early infection based on full-length transcriptome analysis and weighted gene co-expression network analysis. Fish Shellfish Immunol 122, 191–205 (2022).
Zhang, J. et al. Morphological and Molecular Responses of Lateolabrax maculatus Skeletal Muscle Cells to Different Temperatures. Int J Mol Sci 23 (2022).
Tipsmark, C. K., Luckenbach, J. A., Madsen, S. S., Kiilerich, P. & Borski, R. J. Osmoregulation and expression of ion transport proteins and putative claudins in the gill of southern flounder (Paralichthys lethostigma). Comp Biochem Physiol A Mol Integr Physiol 150, 265–73 (2008).
Tse, W. K. F., Au, D. W. T. & Wong, C. K. C. Characterization of ion channel and transporter mRNA expressions in isolated gill chloride and pavement cells of seawater acclimating eels. Biochem Biophys Res Commun 346, 1181–1190 (2006).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–80 (2014).
van Berkum, N.L. et al. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp (2010).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–4 (2008).
Ranallo-Benavidez, T.R., Jaron, K.S. & Schatz, M.C. GenomeScope 2.0 and Smudgeplots: Reference-free proling of polyploid genomes. bioRxiv (2019).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology 31, 1119–1125 (2013).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat Genet 50, 1565–1573 (2018).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845 (2019).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–7 (2007).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–2 (2015).
Feng, Y., JIng, L. & Cheng, X. Study on the karyotype in the Red Drum, Sciaenops ocellatus. 2, 3 (1998).
Xiao Yan, W., Shi, F., wang, JI, S., Zhang & Chang, W., Wu. The Karyotype, Ag-NORs and C-Banding of Sciaenops ocellata. Oceanologia et Limnologia Sinica, 4 (2012).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 13, 1194–1202 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12, 1269–76 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–8 (2005).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–8 (2007).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol 176, 1410–1422 (2018).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–7 (2005).
Neumann, P., Novak, P., Hostakova, N. & Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob DNA 10, 1 (2019).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res 41, D70–82 (2013).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4 10 1–4 10 14 (2009).
Gary, B. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 8 (1999).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Todd, M. L. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25, 9 (1997).
Loman, T. A Novel Method for Predicting Ribosomal RNA Genes in Prokaryotic Genomes. Degree Projects in Bioinformatics BINP30 20161 (2017).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–4 (2005).
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A. & Enright, A. J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34, D140–4 (2006).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–5 (2013).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–44 (2008).
Ian, K. Gene finding in novel genomes. BMC Bioinformatics 5:59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res 44, e89 (2016).
Heckwolf, M. J. et al. Two different epigenetic information channels in wild three-spined sticklebacks are involved in salinity adaptation. Sci Adv 6, eaaz1138 (2020).
Kirubakaran, T. G. et al. A Nanopore Based Chromosome-Level Assembly Representing Atlantic Cod from the Celtic Sea. G3-Genes Genom Genet 10, 2903 (2020).
Ao, J., Mu, Y., Fan, D., Feng, M. & Zhang, S. Genome Sequencing of the Perciform Fish Larimichthys crocea Provides Insights into Molecular and Genetic Mechanisms of Stress Adaptation. PLoS Genet 11, e1005118 (2015).
Ichikawa, K. et al. Centromere evolution and CpG methylation during vertebrate speciation. Nat Commun 8, 1833 (2017).
Shao, C. et al. The genome and transcriptome of Japanese flounder provide insights into flatfish asymmetry. Nat Genet, 3732 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–60 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–5 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–66 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–52 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Consortium, T. G. O. Gene Ontology: tool for the unification of biology. 25, 5 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–62 (2016).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 4 (2000).
Finn, R. D. Pfam: the protein families database. Nucleic Acids Res 42, 9 (2014). e.a.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 8 (1990).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 2 (2015).
Finn, R. D. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res 45, 10 (2017). e.a.
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–9 (2012).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 11, 1650–67 (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biology 15, 550 (2014).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Scardoni, G. et al. Biological network analysis with CentiScaPe: centralities and experimental dataset integration. F1000Res 3, 139 (2014).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–504 (2003).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523 (2019).
Aon, M. A. et al. Untangling Determinants of Enhanced Health and Lifespan through a Multi-omics Approach in Mice. Cell Metab 32, 100–116 e4 (2020).
Vance, J. E. & Vance, D. E. Metabolic insights into phospholipid function using gene-targeted mice. J Biol Chem 280, 10877–80 (2005).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP465111 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP465080 (2023).
Xiao, Y. & Ma, Y. Genbank https://identifiers.org/insdc.gca:GCA_033000465.1 (2023).
Yongshuang, X. Supplementary information for Improved high-quality reference genome of red drum (Sciaenops ocellatus). figshare https://doi.org/10.6084/m9.figshare.24217044.v1 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009).
Acknowledgements
This work was supported by Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB42000000), National Natural Science Foundation of China (No. 42276107; No. 32270472); Biodiversity Conservation Strategy, CAS Strategic Biological Resources Service Network Program (ZSSD-019), Key Deployment Project of Center for Ocean Mega-Science, Chinese Academy of Sciences (COMS2020Q05), National Key Research and Development Program (2022YFC3103600). We also thank Biomarker Technologies (Beijing, China) for their invaluable technical support in this study. We thank anonymous reviewers for helpful comments and suggestions.
Author information
Authors and Affiliations
Contributions
Y.X., J.L. and J.L. conceived the study. Y.X., J.L., Z.X. and J.L. collected the samples, extracted the genomic DNA, and conducted sequencing. Y.X., J.L., J.W. and Y.M. performed bioinformatics analysis. Y.X., J.L. and J.W., wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, Y., Liu, J., Wei, J. et al. Improved high-quality reference genome of red drum facilitates the processes of resistance-related gene exploration. Sci Data 10, 774 (2023). https://doi.org/10.1038/s41597-023-02699-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02699-7
