
Abstract
Articular cartilage has only very limited regenerative capacities in humans. Tissue engineering techniques for cartilage damage repair are limited in the production of hyaline cartilage. Mesenchymal stem/stromal cells (MSCs) are multipotent stem cells and can be differentiated into mature cartilage cells, chondrocytes, which could be used for repairing damaged cartilage. Chondrogenesis is a highly complex, relatively inefficient process lasting over 3 weeks in vitro. Methods: In order to better understand chondrogenic differentiation, especially the commitment phase, we have performed transcriptional profiling of MSC differentiation into chondrocytes from early timepoints starting 15 minutes after induction to 16 hours and fully differentiated chondrocytes at 21 days in triplicates.
Similar content being viewed by others

Background & Summary
Cartilage is a tissue of critical importance in the normal function of the axial skeleton and limb joints, providing the lubricating and shock-absorbing properties of diarthrodial joints and the spine. It also plays a critical role in embryonic limb formation and postnatal growth. Cartilage is a highly specialized avascular and aneural tissue with limited repair capacity1. It consists of distinct populations of chondrocytes and an abundant extracellular matrix rich in collagens and proteoglycans2,3. Traumatic injury of cartilage and degenerative diseases that lead to tissue loss are common and are major causes of disability and loss of mobility. Therapeutic options are limited and there is currently no regenerative treatment that leads to complete restoration of healthy tissue. Thus, there is a compelling need for new therapeutic paradigms and for effective models to understand the regulation of cartilage differentiation and the pathological mechanisms of disease.
Mesenchymal stem/stromal cells (MSCs) represent one such treatment option that has gained much attention in recent years4. In 2018, more than 200 clinical studies using MSCs for cartilage repair were reported5. Compelling data from preclinical studies and from early stage human trials indicate that MSC treatment is a potential disease-modifying approach5,6. This often involves the delivery of undifferentiated MSC preparations by intra-articular injection. Autologous chondrocyte (ACI) transplantation is also commonly used as a treatment for acute focal cartilage injury, with good outcomes. This approach has some disadvantages, namely the need to surgically harvest a tissue biopsy from the articular surface for chondrocyte isolation and the limited expansion capacity of primary cultured chondrocytes. MSCs are multipotent progenitor cells isolated from a wide range of adult tissues7. For therapeutic use the main tissue sources are bone marrow8, adipose tissue8,9, umbilical cord10,11 and placenta12. They are readily expandable through many generations in adherent culture using either planar or bioreactor configurations. They possess a well-described trilineage differentiation propensity and can form cartilage, adipose and bone tissues in vitro and when transplanted in vivo13. The use of chondrocytes derived from MSCs is an attractive option for cartilage repair treatments and overcomes the obstacles associated with using primary chondrocytes14. MSCs are also tolerated as allogeneic therapy, and several advanced therapeutics consisting of allogeneic MSCs have been approved in recent years15,16.
In order to improve the potential therapeutic properties of MSCs for cartilage repair, it is important to understand how and when MSCs commit to chondrogenic differentiation. Of particular interest is the early response to differentiation signals and commitment phase where interventions could be used to increase the number of differentiating cells. Therefore, we examined gene expression during the time-course of chondrogenic differentiation and identified gene regulatory networks (GRNs) that control commitment and differentiation. An obstacle to such studies is the heterogeneity of culture expanded MSC preparations, which consist of mixtures of cells of varying differentiation potential. Therefore, we prepared clonal populations of MSCs from human bone marrow by limiting dilution and obtained a clone with good growth characteristics and clear tri-lineage differentiation potential.
A clonal human MSC line was derived from human bone marrow by limiting dilution cloning as described in the Methods section and validated as described in the Technical Validation section. The clone 1F3 cells, which retained full capacity of chondrogenic, adipogenic and osteogenic differentiation, were induced to differentiate by transferring them from monolayer into 3D culture by gently pelleting the cells and adding TGF-β3. RNA was extracted in triplicates from undifferentiated MSCs in monolayer, undifferentiated MSCs in 3D culture (0 h) and 15 m, 30 m, 1 h, 2 h, 4 h, 8 h, 16 h after adding TGF-β3, and terminally differentiated chondrocytes (21 days after differentiation) (Fig. 1A). Transcriptional profiling in chondrogenesis was previously studied with microarrays in human MSCs17 and with bulk and single-cell RNA-seq in induced pluripotent stem cells (hiPSCs)18. To the best of our knowledge, this is the first bulk RNA-seq transcriptomic study of very early chondrogenesis of bone-marrow derived mesenchymal stem cells.

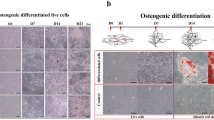
Overview and characterization of a hMSC clone with trilineage differentiation potential. (A) Overview of experimental design. Three replicates for Monolayer MSCs, undifferentiated MSCs in 3D culture (0 h) and 15 m, 30 m, 1 h, 2 h, 4 h, 8 h, 16 h after initiation of differentiation, as well as differentiated chondrocytes (21 days after initiation of differentiation). (B) Schematic of the trilineage differentiation capacity from the bone marrow derived MSC clones from a single donor. (C) The differentiation assays of the clone 1F3 selected for studying the commitment to chondrogenesis. Adipogenic differentiated (i) and control (ii) wells were stained with Oil Red O, with lipid stained vesicles present in the differentiated wells, scale bar 50μm. Osteogenic differentiated (iii) and control (iv) wells were stained with Alizarin Red, and calcium deposition was noted by positive staining in the differentiated wells, scale bar 200μm. Chondrogenic pellets were stained with Safranin O/Fast Green FCF, with positive staining for glycosaminoglycan seen in the differentiated pellets (v) compared to the control pellets (vi), scale bar 100 μM. (D) Surface profile analysis of clonal cells was carried out using flow cytometry. The clonal population was negative for CD3, CD14, CD19, CD34, CD45 and HLA-DR, but positive for the MSC markers CD73, CD90 and CD105. Isotype controls are shown in gray, and the black line represents each antigen. (E) Number or reads sequenced for each sample, for Monolayer MSCs (Mono), undifferentiated MSCs in 3D culture (0 h) and 15 m, 30 m, 1 h, 2 h, 4 h, 8 h, 16 h after initiation of differentiation, as well as differentiated chondrocytes (21 days after initiation of differentiation) replicated in three batches A, B and C. (F) Hierarchical clustering, (G) PCA plot of variance stabilized counts from top 1000 genes. (H) t-Distributed Stochastic Neighbor Embedding with perplexity 5 and max iter 5000.
Methods
Isolation and expansion of bone marrow hMSC clones
hMSCs were isolated from a heparinized bone marrow aspirate harvested from the iliac crest of a healthy 23 year old male volunteer. hMSCs were isolated in vitro by direct plating as described previously19. Cells were plated at a density of 5 × 104 mononuclear cells per cm2 in alpha MEM (Gibco) supplemented with 10% fetal bovine serum (FBS; Hyclone) and 1 ng/mL FGF2 (Peprotech). After five days, when colonies were visible, cells were trypsinized and cloned by limiting dilution into 96 well plates (0.3 cells/well), these clones were termed P1. Wells with single clones were passaged and expanded to P4. The trilineage differentiation potential of the clonal populations were examined, which showed clones with differing propensity for differentiation to the chondrocytic, osteoblastic and adipocytic lineages. A single clone, termed 1F3, with trilineage potential was selected for the study of commitment to chondrogenesis. This clone, as well as having the ability to differentiate to osteoblasts, adipocytes and chondrocytes, also displayed the typical fibroblastic-like morphology and surface marker profile of an MSC.
Characterisation of MSC clones using trilineage differentiation and surface marker expression
MSC clones were characterised by multi-lineage differentiation and flow cytometry for cell surface marker expression. The multi-lineage differentiation capacity of the MSC clones was assessed by their differentiation to the classical MSC lineages i.e., chondrogenic, osteogenic and adipogenic using established protocols19.
Chondrogenic potential was examined using a 3-D pellet culture system, where, 2.5 × 105 cells were pelleted at 100 xg for 5 minutes in complete chondrogenic medium (CCM) consisting of Dulbecco’s Modified Eagle Serum (DMEM, 4.5 g/L glucose), 2 mM glutamine, 100 mM dexamethasone, 50 μg/mL ascorbic acid, 40 μg/mL L-Proline, 1% ITS + (Insulin, Transferrin, Selenium) (Corning), 1 mM sodium pyruvate and penicillin-streptomycin (100 U/mL), supplemented with 10 ng/mL TGF-β3 (Peprotech). Control pellets were incubated without TGF-β3 incomplete chondrogenic medium (ICM). After 21 days, pellets were fixed in 10% neutral buffered formalin, and processed for histology in a Leica ASP300S tissue processor and embedded in paraffin. Sections were stained with Safranin O and Fast Green FCF and imaged with an Olympus BX43 microscope.
For osteogenesis, cells were seeded in culture medium and when the monolayer reached 90% confluence, the medium was replaced with osteogenic medium containing DMEM (1 g/L glucose; Sigma Aldrich), 2 mM l-glutamine, 100 nM dexamethasone, 100 μM ascorbic acid, 10 mM β-glycerophosphate, 10% FBS (Hyclone) and penicillin-streptomycin (100 U/ml). Medium was replaced every 3–4 days for up to 14 days, when monolayers were fixed with 10% ice cold methanol, then stained with 2% Alizarin Red and imaged using an Olympus IX71 microscope.
Adipogenesis was assessed by incubating confluent cultures in adipogenic induction medium comprising DMEM (4.5 g/L glucose), 2 mM l-glutamine, 10% FBS (Hyclone), 1 μM dexamethasone, 10 μg/mL insulin (Roche), 200 μM indomethacin, 500 μM 3-isolbutyl-1-methylxanthine and penicillin-streptomycin (100 U/mL). After 3 days, the culture was transferred to adipogenic maintenance medium comprising DMEM (4.5 g/L glucose), 10% FBS (Hyclone), 10 μg/mL insulin (Roche) and penicillin-streptomycin (100 U/mL) for 1 day. This cycle was repeated 3 times after which the cells were maintained in adipogenic maintenance medium for a further 5 days. Cultures were fixed in 10% neutral buffer formalin and stained with Oil Red O before imaging using an inverted Olympus IX71 microscope.
Surface marker expression of MSC clones was carried out by flow cytometry using the BD FACS Canto II flow cytometer (BD Biosciences) using antibodies against CD3, CD14, CD19, CD34, CD45, HLA-DR and the MSC positive markers CD73, CD90 and CD105 (BD Biosciences) as described previously20. Post-acquisition analysis was carried out using the FlowJo software (Treestar Inc.).
Study of the commitment towards chondrogenesis
To examine the commitment towards chondrogenesis, clonal cell cultures were switched to a culture medium containing 1% FBS twelve hours prior to chondrogenic induction. Chondrogenic differentiation was carried out in a pellet culture format as described above. Three pellets were set up for each time point. Pellets from the 0 h time point remained in ICM, while pellets at 15 min, 30 min, 1, 2, 4, 8 and 16 h time points were cultured in CCM. At each time point the 3 pellets were combined into 1 tube, the pooled pellets were washed in PBS and snapped frozen in liquid nitrogen. Clonal cells in monolayer and 21d chondrogenic pellets were included as controls. The time course experiment was carried out three times and each time course served as a technical replica for the experiment.
RNA extraction and sequencing
Total RNA was extracted using an miRNeasy Mini kit (QIAGEN). For day 21 samples, due to the amount of matrix deposition around the chondrocytes in this late time point pellet, mechanical homogenisation by pulverizing in a chilled steel mortar and pestle was required prior to RNA extraction21. The concentration and purity of the isolated RNA was determined using the Nanodrop ND-1000 (Nanodrop Technologies). RNA integrity was measured using the 2100 Bioanalyzer (Agilent Technologies). Bioanalyzer and Nanodrop results were deposited on Zenodo as lab_qc.zip22. 400 ng of total RNA was included in each sequencing reaction.
Poly(A)+ enrichment to purify mRNA from each sample was performed by hybridization of the RNA with poly(T) oligomers. The mRNA was then converted to cDNA and fragmented, creating cDNA library fragments of approximately 250nt in size. Sequencing adaptors were then added to each cDNA library fragment. The libraries were sequenced on an Illumina GAIIx using TruSeq Single read Cluster Generation Kit v5 and TruSeq Single read 36 cycle Sequencing kit v5. The sequencing mode was 35 nt single-read plus 7 nt for indices. The raw reads were aligned to Human Gencode23 (v23/h38.p3) reference genome with STAR aligner24 (v2.5.0a) and summarized with featureCounts25 (v.1.6.2).
Ethics approval and consent to participate
All procedures were approved by the Clinical Research Ethical Committee of Galway University Hospital and by the National University of Ireland Galway Research Ethics Committee, including written consent to participate and publish sequenced data.
Data Records
Availability of data and materials
Transcriptional profiling of undifferentiated human MSCs (Mono), MSCs in 3D culture (0 h), early differentiation time-points 15 m, 30 m, 1 h, 2 h, 4 h, 8 h, 16 h after induction with TGF-β3 and hyaluronic acid, and fully differentiated chondrocytes (21 days after induction) using RNA-seq in triplicates of clones was performed. Raw sequencing data (fastq), sample information (MAGE-TAB format) and summarized counts (tab-delimited text file) have been submitted to BioStudies under accession number E-MTAB-1047626. MultiQC of FastQC and alignment statistics, PCA and differential gene expression analysis with DESeq227 and edgeR28, time-course clustering Mfuzz29, BigWig files, function annotation and functional analysis with Ingenuity Pathway Analysis and Cluster Profiler30 was deposited on Zenodo22.
Technical Validation
Selection of 1F3 clone
In total 21 clones were isolated from one donor, of which 11 had trilineage capacity (Fig. 1B). The selected clone 1F3 retained the full capacity of MSCs for chondrogenic, adipogenic and osteogenic differentiation (Fig. 1C). Surface marker expression of the MSC clone was carried out by flow cytometry using PE labelled antibodies (BD Biosciences) against CD3, CD14, CD19, CD34, CD45, HLA-DR and the MSC positive markers CD73, CD90 and CD105 as described previously19. Antibody details are supplied in supplementary22. Cells were washed and blocked in PBS with 2% FBS prior to incubation for 30 minutes with antibodies, diluted to the recommended concentrations according to the manufacturer’s instructions. Following washing, Sytox Red (Invitrogen), a stain for dead cells was added prior to acquisition of the samples on a BD FACS Canto II (BD Biosciences), where the live single cell population was analyzed. Post-acquisition analysis was carried out using the FlowJo software (Treestar Inc.). Flow cytometric analysis of cell surface antigens showed that the clones were positive for MSC markers CD105, CD90 and CD73 and negative for hematopoietic markers CD45, CD34, CD19, CD3 and CD14, with minor expression of HLA-DR (Fig. 1D).
Bioinformatics Validation of Sequencing data
The raw reads were aligned to Human Gencode23 (v23/h38.p3) reference genome with STAR aligner24 (v2.5.0a) and summarized with featureCounts25 (v.1.6.2) and are available in BioStudies26. The median library size is 21.52 million reads (Fig. 1E). The three differentiated (21-days) samples have 13.69 million reads on average, due to technical challenges of RNA extraction from differentiated chondrocytes. Hierarchical clustering (Fig. 1F), Principal component analysis (PCA) (Fig. 1G) of the top thousand variable genes as well t-SNE confirming that the experimental conditions are very reproducible (Fig. 1H).
Usage Notes
Functional annotation, differential gene expression analysis, time-course clustering and upstream regulator prediction workflow can be found at Zenodo22.
Code availability
A compiled protocol and supplementary files and information are deposited as ‘report.zip’ at Zenodo22. The index.html links to the analysis report and compiled R markdown file. The raw code and source annotations are deposited as ‘analysis.zip’ in the same repository.
References
Sherwood, J. C., Bertrand, J., Eldridge, S. E. & Dell’Accio, F. Cellular and molecular mechanisms of cartilage damage and repair. Drug Discov. Today 19, 1172–1177 (2014).
Sophia Fox, A. J., Bedi, A. & Rodeo, S. A. The basic science of articular cartilage: structure, composition, and function. Sports Health 1, 461–468 (2009).
Becerra, J. et al. Articular Cartilage: Structure and Regeneration. Tissue Eng. Part B Rev. 16, 617–627 (2010).
Zha, K. et al. Recent Developed Strategies for Enhancing Chondrogenic Differentiation of MSC: Impact on MSC-Based Therapy for Cartilage Regeneration. Stem Cells Int. 2021 (2021).
Negoro, T., Takagaki, Y., Okura, H. & Matsuyama, A. Trends in clinical trials for articular cartilage repair by cell therapy. NPJ Regen Med 3, 17 (2018).
Murphy, J. M., Fink, D. J., Hunziker, E. B. & Barry, F. P. Stem cell therapy in a caprine model of osteoarthritis. Arthritis Rheum. 48, 3464–3474 (2003).
Colombini, A. et al. Mesenchymal stem cells in the treatment of articular cartilage degeneration: New biological insights for an old-timer cell. Cytotherapy 21, 1179–1197 (2019).
Prockop, D. J. Marrow Stromal Cells as Stem Cells for Nonhematopoietic Tissues. Science 276, 71–74 (1997).
Zuk, P. A. et al. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 13, 4279–4295 (2002).
Romanov, Y. A., Svintsitskaya, V. A. & Smirnov, V. N. Searching for alternative sources of postnatal human mesenchymal stem cells: candidate MSC-like cells from umbilical cord. Stem Cells 21, 105–110 (2003).
Wang, H.-S. et al. Mesenchymal stem cells in the Wharton’s jelly of the human umbilical cord. Stem Cells 22, 1330–1337 (2004).
Fukuchi, Y. et al. Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells 22, 649–658 (2004).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006).
Pelttari, K., Steck, E. & Richter, W. The use of mesenchymal stem cells for chondrogenesis. Injury 39, 58–65 (2008).
Barry, F. MSC therapy for osteoarthritis: An unfinished story. J. Orthop. Res. 37, 1229–1235 (2019).
Hoogduijn, M. J. & Lombardo, E. Mesenchymal Stromal Cells Anno 2019: Dawn of the Therapeutic Era? Concise Review. Stem Cells Transl. Med. 8, 1126–1134 (2019).
Sotoca, A. M., Weber, M. & van Zoelen, E. J. J. Gene expression regulation underlying osteo-, adipo-, and chondro-genic lineage commitment of human mesenchymal stem cells. Medical Advancements in (2013).
Wu, C. L., Dicks, A., Steward, N., Tang, R. & Katz, D. B. Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nature (2021).
Murphy, J. M. et al. Reduced chondrogenic and adipogenic activity of mesenchymal stem cells from patients with advanced osteoarthritis. Arthritis Rheum. 46, 704–713 (2002).
Ferro, F. et al. Survival/adaptation of bone marrow-derived mesenchymal stem cells after long-term starvation through selective processes. Stem Cells 37, 813–827 (2019).
Mallein-Gerin, F. & Gouttenoire, J. RNA extraction from cartilage. Methods Mol. Med. 100, 101–104 (2004).
Schwarzl, T. & Kolch, W. Analysis and Report for ‘Transcriptional profiling of early differentiation of primary human mesenchymal stem cells into chondrocytes’. Zenodo https://doi.org/10.5281/zenodo.8079809 (2022).
Hancock, J.M., Zvelebil, M.J., Blanco, E. and Abril, J.F. GENCODE. In Dictionary of Bioinformatics and Computational Biology (eds J.M. Hancock and M.J. Zvelebil, 2014) https://doi.org/10.1002/9780471650126.dob0915.
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Schwarzl, T. et al. Transcriptional profiling of early differentiation of primary human mesenchymal stem cells into chondrocytes E-MTAB-10476. ArrayExpress https://identifiers.org/arrayexpress:E-MTAB-10476 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Kumar, L. & Futschik, M. E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS 16, 284–287 (2012).
Acknowledgements
We thank University College Dublin Conway Institute Core Technology Facilities for performing RNA sequencing. We thank Thileepan Sekaran for support with the summarization with featureCounts. This work was supported by the Science Foundation Ireland under Grants No. 06/CE/B1129 and 18/SPP/3522.
Author information
Authors and Affiliations
Contributions
T.S. performed the bioinformatic analysis. T.S. wrote the manuscript. A.K. and G.S. contributed sections to the manuscript. W.K., D.G.H. and A.K. edited the manuscript. A.K. did the I.P.A. analysis of cluster upstream regulators. F.B. designed the experiment. E.C., An.K., G.S. and M.M. performed the experimental work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwarzl, T., Keogh, A., Shaw, G. et al. Transcriptional profiling of early differentiation of primary human mesenchymal stem cells into chondrocytes. Sci Data 10, 758 (2023). https://doi.org/10.1038/s41597-023-02686-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02686-y
