
Abstract
The East Sea, also known as the Sea of Japan, is a marginal sea located in the western Pacific Ocean, displaying comparable characteristics to Earth’s oceans, thereby meriting its recognition as a “miniature ocean”. The East Sea exhibits a range of annually-recurring biogeochemical features in accordance with seasonal fluctuations, such as phytoplankton blooms during the spring and autumn seasons. Despite ongoing monitoring efforts focused on water quality and physicochemical parameters, the investigation of prokaryotic assemblages in the East Sea, encompassing seasonal variations, has been infrequently pursued. Here, we present a monthly time-series metagenomic dataset spanning a one-year period in 2009, obtained from surface (10 m) seawater samples collected off the coast of the East Sea. The dataset encompasses 12 metagenomes, amounting 195 Gbp, with 14.73–22.52 Gbp per sample. This dataset is accompanied by concurrently measured physicochemical parameters. Our anticipation is that these metagenomes will facilitate extensive investigations aimed at elucidating various aspects of the marine microbial ecosystems in the East Sea.
Similar content being viewed by others


Background & Summary
Metagenomics has emerged as a fundamental approach in marine environmental studies, deciphering the intricacies and diversity of microbial communities in the oceans and their environmental interplay1,2,3,4. This technology has facilitated the discovery of hitherto unknown microbes5,6,7, genes8,9,10, and metabolic pathways11,12, thereby considerably enriching our understanding of marine biodiversity and ecosystem function. Additionally, metagenomic techniques present opportunities for new biotechnological discoveries, such as enzyme development for industry and the identification of bioactive compound sources13,14,15. The continuous evolution of metagenomic tools can substantially augment our comprehension of Earth’s ecosystems, necessitating the generation and efficient exploitation of purpose-aligned metagenomic data.
The East Sea, which is also referred to as the Sea of Japan, is a semi-enclosed marginal sea situated in the western Pacific Ocean, and is colloquially termed a “miniature ocean” owing to its resemblance to the global oceans16. One of the prominent features of the East Sea is the Tsushima Warm Current (TWC), which originates from the south and is a major driver in shaping the region’s oceanic circulation by modulating water temperature, salinity, and nutrient dispersion. The substantial nutrient influx via these currents, in tandem with coastal upwelling17,18, results in high primary productivity, especially associated with the annual cycle of spring (April-June) and autumn (October-November) phytoplankton blooms19,20,21. Such recurrent biogeochemical fluctuations necessitate regular assessments to understand their impact on the microbial ecology of the East Sea, as shown by studies in the North Sea22.
Numerous long-term studies from the East Sea have been undertaken via programs including the Circulation Research in the East Asian Marginal Sea (CREAMS), CTD station operations23,24 or satellite colour measurements20,25. To date, however, there appears to be a lacuna in studies specifically focusing on the monthly variation of microbial community structure in the East Sea, which are indicative of seasonal changes in environmental parameters such as chlorophyll a concentration. Previous microbial investigations in the East Sea have primarily concentrated on the deep-sea sediments or methane hydrate-containing sediments26,27,28,29. In addition, a metagenomic study has been undertaken to assess the influence of environmental determinants on the spatial distribution of pelagic bacteria in the East Sea, albeit limited to a bimonthly scale during the summer and winter months30.
In this study, we present a one-year (January to December 2009) monthly metagenomic dataset derived from the East Sea’s coastal waters. Seawater samples were filtered using a 0.2 µm pore-size membrane and subsequently cryopreserved at −80 °C until DNA extraction, followed by sequencing via the Illumina HiSeq platform. The physicochemical characteristics of the water samples were concurrently measured to infer the environmental factors influencing the microbial community. The schematic diagram illustrating the methodology used for generating this dataset is presented in Fig. 1.

Schematic diagram of the processing of seawater samples.
Our selected sampling locations are of considerable scientific import due to the successful in situ isolation of a plethora of bacterial strains belonging to major marine bacterial clades, including SAR11, oligotrophic marine gammaproteobacteria (e.g., SAR92, OM60), OM43, and SAR11631,32,33. Therefore, the creation of this monthly metagenomic repository can offer an asset for investigating prokaryotic assemblages of temperate coastal seas through both culture-dependent and culture-independent methodologies. Furthermore, metagenomic analyses may unveil previously uncultured microbial species and suggest potential cultivation strategies. This exhaustive insight into the microbial community of this “miniature ocean” holds promise for fostering a deeper understanding of global marine ecosystems.
Methods
Sampling process
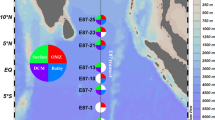
Seawater samples were collected monthly off the coast of the East Sea, in proximity to Sokcho, Korea, throughout the year 2009. Sampling was executed approximately 8 km from Dongmyeong Port (Fig. 2 and Table 1), with the exact location of sampling stations subject to minor variations due to atmospheric conditions. Approximately 10 litres of surface seawater samples were collected from a depth of 10 m using a Niskin sampler (General Oceanics, Inc., USA) and were transported to the laboratory in an ice-cooled box. The water samples (6 litres; 6 replicates of each 1 litre) were filtered through 0.2 μm pore-size polyethersulfone membrane filter (47 mm in diameter, Supor®, Pall, USA) for DNA extraction. Additionally, 1 litre was filtered using a 47 mm GF/F glass-fiber filter (Whatman, USA) to analyse chlorophyll a. All filters were stored at −80 °C until further processing. The residual volume was filtered employing a 0.45 μm pore-size cellulose ester membrane filter (Advantec, Japan), aliquoted into 50 ml conical tubes (Falcon, USA), and preserved at −80 °C, to be later used for the analysis of environmental parameters including dissolved inorganic ions (ammonium, nitrite + nitrate, phosphate, and silicate). Temperature and salinity of the water samples were measured onboard using a YSI 30 (YSI Inc., USA). The total cell count was conducted using epifluorescence microscopy (Nikon 80i, Nikon, Japan), enumerating DAPI-stained cells (Table 2).

Map of the sampling stations. Sampling stations of each month are indicated as blue dots with three-letter abbreviations of months. Inset at the upper left shows the approximate location of the sampling stations in the East Sea.
Biogeochemical analyses
Chlorophyll a was extracted from GF/F glass-fiber filters using 90% aqueous acetone (v/v) at 4 °C overnight. The extraction solution was centrifuged for 10 min at 2,000 rpm, and the supernatants were analysed via a fluorometer (10 AU, Turner Designs, USA). Concentrations of inorganic nutrients, including NO2−, NO3−, NH4+, PO43−, and SiO2, were determined employing a QuAAtro Microflow Analyzer (SEAL Analytical, UK). The values obtained are graphically represented in Fig. 3 and tabulated in Table 2.

Physicochemical parameters of seawater samples collected monthly.
DNA extraction and metagenome sequencing
DNA was extracted from membrane filters using a protocol based on manual cell lysis, followed by purification with the DNeasy Blood & Tissue Kit (Qiagen, Germany). The membranes were situated inside a 5 ml tube with the sample-filtered side inward. Subsequent to the addition of 1 ml of cell lysis buffer (20 mM EDTA, 50 mM Tris, 400 mM NaCl, and 0.75 M sucrose) and 5 μl of lysozyme solution (10 mg mL−1 in 10 mM Tris-Cl (pH 8.0)), tubes were incubated for 30 min at 37 °C in a horizontal orientation with a rotation speed of 5 rpm in a hybridization oven. Following this, proteinase K at a final concentration of 0.2 mg mL−1 and sodium dodecyl sulfate at a final concentration of 1% were introduced, and the tubes were further incubated at 55 °C overnight with rotation in a hybridization oven. After incubation, RNase A (200 μg mL−1), 1 mL of AL buffer (DNeasy Blood & Tissue Kit, Qiagen), and 70% ethanol were sequentially added to the tubes with appropriate incubation times. The manufacturer’s instructions of DNeasy Blood & Tissue Kit were adhered to from the stage of transferring the lysis mixture to the DNeasy Mini spin column. The quality and quantity of the extracted DNA were assessed using electrophoresis with 1% of agarose gel, Nanodrop ND-1000 (Thermo Fischer Scientific, USA), and Qubit 2.0 Fluorometer (Life Technologies, US) employing the Qubit® dsDNA Assay Kit. Metagenome sequencing was performed at Theragen Etex Inc. (Suwon, Korea). The Truseq library preparation kits with default library linkers and adaptors were used to generate sequencing libraries. The libraries were sequenced on an Illumina HiSeq 2500 platform, producing 250 bp paired-end reads.
Taxonomic profiling of metagenomic reads
Raw sequence reads were decontaminated by adapter removal and quality trimming using BBDuk (v39.01) with the following parameters: ktrim = r, k = 23, mink = 11, hdist = 1, tpe, tbo, ftm = 5, qtrim = rl, trimq = 10, minlen = 100. Subsequently, the taxonomic profiling of these metagenomic reads was performed against a customized GTDB database (R207) generated by Struo234 (http://ftp.tue.mpg.de/ebio/projects/struo2/GTDB_release207/). Taxonomic classification and species abundance estimation were performed using Kraken2 (v2.1.3) and Bracken (v2.7)35. The organization of the output report file was accomplished using Pavian36 (https://fbreitwieser.shinyapps.io/pavian/). Finally, the resulting species abundance information was visualized using the R package ‘tidyverse’.
Technical Validation
The assessment of quality scores for the raw reads of the 12 metagenomes was performed using FastQC (v0.10.1). The results show that ~91.88% and ~79.60% of the bases have quality scores of ≥20 and ≥30, respectively, indicating that sequencing was performed successfully (Fig. 4). The distribution of per-read quality scores across the 12 metagenomes was similar, further indicating no quality issues (Fig. 4). Consistent with the characteristics of the Illumina sequencing technology, the forward reads exhibited higher quality compared to the reverse reads (Fig. 4). A succinct taxonomic profiling analysis was then conducted to ascertain the suitability of the generated data for subsequent metagenomic analysis (Fig. 5). The taxonomic composition revealed a prominent dominance of Pelagibacterales (26.0%; a median value over a 12-month period), followed by Flavobacteriales (12.6%), SAR86 (7.7%), Pseudomonadales (5.5%), and Rhodobacterales (5.3%). These taxa are typically known for their prevalence in the ocean1,4. This observed pattern also aligns with the microbial community structure derived from culture-independent investigations conducted on a seawater sample collected from the same research station31.

Distribution of per sequence mean quality scores of the 12 metagenomes.

Taxonomic profiling of metagenomic reads retrieved from the East Sea. A total of 34 orders are presented that ranked in the top 30 by maximum or median among the 12 metagenome samples.
Code availability
FastQC (v0.10.1) was used to check the quality of the raw data. No other code or software was used, as the original sequences were submitted.
References
Sunagawa, S. et al. Structure and function of the global ocean microbiome. Science 348, 1261359, https://doi.org/10.1126/science.1261359 (2015).
Acinas, S. G. et al. Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Commun. Biol. 4, 604, https://doi.org/10.1038/s42003-021-02112-2 (2021).
Tringe, S. G. et al. Comparative metagenomics of microbial communities. Science 308, 554–557, https://doi.org/10.1126/science.1107851 (2005).
Venter, J. C. et al. Environmental genome shotgun sequencing of the Sargasso Sea. science 304, 66–74, https://doi.org/10.1126/science.1093857 (2004).
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 1–6, https://doi.org/10.1038/nmicrobiol.2016.48 (2016).
Spang, A. et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179, https://doi.org/10.1038/nature14447 (2015).
Lewis, W. H., Tahon, G., Geesink, P., Sousa, D. Z. & Ettema, T. J. Innovations to culturing the uncultured microbial majority. Nat. Rev. Microbiol. 19, 225–240, https://doi.org/10.1038/s41579-020-00458-8 (2021).
Ahlgren, N. A., Fuchsman, C. A., Rocap, G. & Fuhrman, J. A. Discovery of several novel, widespread, and ecologically distinct marine Thaumarchaeota viruses that encode amoC nitrification genes. ISME J. 13, 618–631, https://doi.org/10.1038/s41396-018-0289-4 (2018).
He, S. et al. Diversity, distribution, and expression of opsin genes in freshwater lakes. Mol. Ecol. 32, 2798–2817, https://doi.org/10.1111/mec.16891 (2023).
Béja, O. et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906, https://doi.org/10.1126/science.289.5486.1902 (2000).
McKay, L. J. et al. Activity-based, genome-resolved metagenomics uncovers key populations and pathways involved in subsurface conversions of coal to methane. ISME J. 16, 915–926, https://doi.org/10.1038/s41396-021-01139-x (2022).
Hallam, S. J. et al. Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305, 1457–1462, https://doi.org/10.1126/science.1100025 (2004).
Kennedy, J., Marchesi, J. R. & Dobson, A. D. Marine metagenomics: strategies for the discovery of novel enzymes with biotechnological applications from marine environments. Microb. Cell Factories 7, 1–8, https://doi.org/10.1186/1475-2859-7-27 (2008).
Schloss, P. D. & Handelsman, J. Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 14, 303–310, https://doi.org/10.1016/S0958-1669(03)00067-3 (2003).
Lee, H. S. et al. Approaches for novel enzyme discovery from marine environments. Curr. Opin. Biotechnol. 21, 353–357, https://doi.org/10.1016/j.copbio.2010.01.015 (2010).
Ichiye, T. Some problems of circulation and hydrography of the Japan Sea and the Tsushima Current. Ocean Hydrodynamics of the Japan and East China Seas in Elsevier Oceanography Series. Vol. 39, 15–54, https://doi.org/10.1016/S0422-9894(08)70289-7 (Elsevier, 1984).
Onitsuka, G., Yanagi, T. & Yoon, J. H. A numerical study on nutrient sources in the surface layer of the Japan Sea using a coupled physical‐ecosystem model. J. Geophys. Res. Oceans 112, C5, https://doi.org/10.1029/2006JC003981 (2007).
Yoo, S. & Park, J. Why is the southwest the most productive region of the East Sea/Sea of Japan? J. Mar. Syst. 78, 301–315, https://doi.org/10.1016/j.jmarsys.2009.02.014 (2009).
Kim, S.-W., Saitoh, S.-i., Ishizaka, J., Isoda, Y. & Kishino, M. Temporal and spatial variability of phytoplankton pigment concentrations in the Japan Sea derived from CZCS images. J. Oceanogr. 56, 527–538, https://doi.org/10.1023/A:1011148910779 (2000).
Wang, D. et al. Satellite-detected phytoplankton blooms in the Japan/East Sea during the past two decades: magnitude and timing. Front. Mar. Sci. 2503, https://doi.org/10.3389/fmars.2022.1065066 (2022).
Lee, J.-Y. et al. Spatial and temporal variability in the pelagic ecosystem of the East Sea (Sea of Japan): a review. J. Mar. Syst. 78, 288–300, https://doi.org/10.1016/j.jmarsys.2009.02.013 (2009).
Teeling, H. et al. Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. Elife 5, e11888, https://doi.org/10.7554/eLife.11888 (2016).
Kim, K. et al. Water masses and decadal variability in the East Sea (Sea of Japan). Prog. Oceanogr. 61, 157–174, https://doi.org/10.1016/j.pocean.2004.06.003 (2004).
Kim, K.-R. & Kim, K. What is happening in the East Sea (Japan Sea)?: Recent chemical observations during CREAMS 93-96. J. Kor. Soc. Oceanogr. 31, 164–172 (1996).
Park, K.-A., Park, J.-E. & Kang, C.-K. Satellite-Observed Chlorophyll-a Concentration Variability in the East Sea (Japan Sea): Seasonal Cycle, Long-Term Trend, and Response to Climate Index. Front. Mar. Sci. 9, 807570, https://doi.org/10.3389/fmars.2022.807570 (2022).
Lee, J.-W. et al. Microbial community structures of methane hydrate-bearing sediments in the Ulleung Basin, East Sea of Korea. Mar. Pet. Geol. 47, 136–146, https://doi.org/10.1016/j.marpetgeo.2013.06.002 (2013).
Jeong, I. S. et al. Vertical profile of bacterial community in the sediment of Ulleung Basin: implication of the presence of methane-driven community in Microorganisms In Industry And Environment: From Scientific and Industrial Research to Consumer Products. (Mendez-vilas, A. Ed.) 219-226, (World Scientific, 2011).
Shin, J.-H., Nam, J.-H., Lee, J.-W. & Lee, D.-H. Characteristics of Microbial Community Structures of the Methane Hydrate Sediments in the Ulleung Basin, East Sea of Korea. Korean J. Microbiol. 50, 191–200, https://doi.org/10.7845/kjm.2014.4033 (2014).
Cho, H. et al. Microbial community structure associated with biogeochemical processes in the sulfate–methane transition zone (SMTZ) of gas-hydrate-bearing sediment of the Ulleung Basin, East Sea. Geomicrobiol. J. 34, 207–219, https://doi.org/10.1080/01490451.2016.1159767 (2017).
Kim, H.-R., Lim, J.-H., Kim, J.-H., Thangaraj, S. & Kim, I.-N. Physical Process Controlling the Surface Bacterial Community Composition in the Ulleung Basin of East Sea. Front. Mar. Sci. 9, 841492, https://doi.org/10.3389/fmars.2022.841492 (2022).
Yang, S.-J., Kang, I. & Cho, J.-C. Expansion of cultured bacterial diversity by large-scale dilution-to-extinction culturing from a single seawater sample. Microb. Ecol. 71, 29–43, https://doi.org/10.1007/s00248-015-0695-3 (2016).
Song, J., Oh, H.-M. & Cho, J.-C. Improved culturability of SAR11 strains in dilution-to-extinction culturing from the East Sea, West Pacific Ocean. FEMS Microbiol. Lett. 295, 141–147, https://doi.org/10.1111/j.1574-6968.2009.01623.x (2009).
Li, S.-H. et al. Halioglobus maricola sp. nov., isolated from coastal seawater. Int. J. Syst. Evol. Microbiol. 70, 1868–1875, https://doi.org/10.1099/ijsem.0.003985 (2020).
Youngblut, N. D. & Ley, R. E. Struo2: efficient metagenome profiling database construction for ever-expanding microbial genome datasets. PeerJ. 9, e12198, https://doi.org/10.7717/peerj.12198 (2021).
Lu, J. et al. Metagenome analysis using the Kraken software suite. Nat. Protoc. 17, 2815–2839, https://doi.org/10.1038/s41596-022-00738-y (2022).
Breitwieser, F. P. & Salzberg, S. L. Pavian: interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 36, 1303–1304, https://doi.org/10.1093/bioinformatics/btz715 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP396155 (2022).
Acknowledgements
This research was supported by High Seas Bioresources Program of Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (KIMST-20210646), by the Mid-Career Research Program (NRF-2022R1A2C3008502), and by the Basic Science Research Program (NRF-2022R1A6A3A01087360) through the National Research Foundation (NRF) funded by the Ministry of Sciences and Information and Communications Technology, Korea.
Author information
Authors and Affiliations
Contributions
S.-J.Y. designed a project and conducted the monthly sampling. Y.L. and I.K. generated and processed the data. Y.L., I.K. and J.-C.C. wrote and revised the manuscript. I.K. and J.-C.C. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, Y., Yang, SJ., Kang, I. et al. Metagenomic data from surface seawater of the east coast of South Korea. Sci Data 10, 647 (2023). https://doi.org/10.1038/s41597-023-02556-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02556-7
