
Abstract
The darkbarbel catfish (Pelteobagrus vachelli), an economically important aquaculture species in China, is extensively employed in hybrid yellow catfish production due to its superior growth rate. However, information on its genome has been limited, constraining further genetic studies and breeding programs. Leveraging the power of PacBio long-read sequencing and Hi-C technologies, we present a high-quality, chromosome-level genome assembly for the darkbarbel catfish. The resulting assembly spans 692.10 Mb, with an impressive 99.9% distribution over 26 chromosomes. The contig N50 and scaffold N50 are 13.30 Mb and 27.55 Mb, respectively. The genome is predicted to contain 22,109 protein-coding genes, with 96.1% having functional annotations. Repeat elements account for approximately 35.79% of the genomic landscape. The completeness of darkbarbel catfish genome assembly is highlighted by a BUSCO score of 99.07%. This high-quality genome assembly provides a critical resource for future hybrid catfish breeding, comparative genomics, and evolutionary studies in catfish and other related species.
Similar content being viewed by others

Background & Summary
Siluriformes, better known as catfishes, constitute a significant portion of the teleost orders, making up approximately 11% of all species. With 39 families and around 4,094 species documented to date, this order is among the largest in existence1. Owing to their delectable meat, minimal intermuscular bone, and impressive feed conversion ratio, catfishes have risen to become one of the top three farmed fish and shellfish, boasting a production of 5,519 kilotons in 2017 alone2,3.
Yellow catfish (Pelteobagrus fulvidraco) is an important aquaculture fish species in China, the production of which was about 565 thousand tons in 20204. Darkbarbel catfish (Pelteobagrus vachelli), a close relative of yellow catfish within the family Bagridae, has a faster growth rate than yellow catfish5. Recently, hybrid yellow catfish (P. fulvidraco♀ × P. vachelli♂) has been widely cultured in China due to its faster growth rate and better emergence rate6. Unfortunately, specific research on the darkbarbel catfish genome proved difficult to access, but the general principles of genomics in aquaculture suggest that having a high-quality reference genome for this species will have significant benefits for breeding programs and other genetic studies. The insights gained from the darkbarbel catfish genome can also support the successful production of the hybrid yellow catfish, by contributing to a better understanding of their genetic makeup and the genetic factors influencing their advantageous traits.
In this research, we have employed a combination of PacBio long-read sequencing and Hi-C technology to generate a high-quality, chromosome-level assembly of the darkbarbel catfish genome. With the development of this high-quality reference genome, we foresee a significant propulsion in the field of population genetics and the identification of functional genes associated with critical economic traits in the darkbarbel catfish. The elucidation of these genomic underpinnings is expected to provide deeper insights into the hybrid vigor observed in catfish hybrids, thereby contributing to the optimization of hybrid breeding strategies.
Methods
Sample collection and sequencing
An adult female darkbarbel catfish was collected from the Yangtze River in Wuhan, Hubei, China. High-molecular weight (HMW) genomic DNA was extracted from muscle for Illumina sequencing and PacBio SMRT sequencing. The quality and quantity of the extracted DNA was assessed using standard agarose gel electrophoresis and a Qubit fluorometer (Thermo Fisher Scientific, USA).
For Illumina sequencing, the genomic DNA was randomly sheared to ~350 bp fragments, and a paired-end genomic library was prepared following the manufacturer’s protocol. Then, the library was sequenced on an Illumina HiSeq X-Ten platform using a paired-end 150 bp layout. For PacBio sequencing, the genomic DNA was used to construct SMRTbell libraries following the manufacturer’s protocol. After that, the libraries were sequencing on a PacBio Sequel platform with SMRT technology. Finally, we generated 96.33 Gb Illumina short-read data and 109.76 Gb of raw PacBio continuous long reads (CLR) with an average read length of 13.8 kb and a N50 read length of 22.4 kb (Table 1).
For genome scaffolding, a Hi-C library was prepared using blood sample from the same darkbarbel catfish used for genomic DNA sequencing. The Hi-C library construction, including cell crosslinking, cell lysis, chromatin digestion, biotin labelling, proximal chromatin DNA ligation and DNA purification, was performed as previously described7, and the resulting Hi-C library was then subjected to paired-end sequencing with 150 bp read lengths on an Illumina HiSeq X-Ten platform. As a result, 89.12 Gb of Hi-C read data was generated (Table 1).
To aid in genome annotation, total RNA was extracted from multiple tissues, including spleen, kidney, brain, muscle, ovary and liver, and the quality were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The mixed RNA sample was used to construct a cDNA library using the TruSeq Stranded mRNA Library Prep Kit (Illumina, USA) following the manufacturer’s protocol. The library was then sequenced on an Illumina HiSeq X-Ten platform using a paired-end 150 bp layout, and 13.11 Gb of data was obtained (Table 1).
Genome assembly and polishing
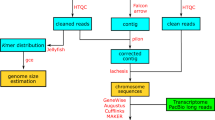
To assemble the genome, we utilized two different assemblers: Wtdbg2 v2.58 and Flye v2.99 (Fig. 1a). The assembly generated by each assembler with default parameters was then polished using Arrow, a consensus algorithm that can generate highly accurate consensus sequences from PacBio subreads. The two polished assemblies were then merged using Quickmerge10, a tool that combine multiple genome assemblies into a single consensus assembly. The resulting merged assembly was then polished twice using two rounds of Arrow and two rounds of NextPolish with default parameters (Fig. 1a). We used PacBio subreads for Arrow and Illumina short reads for NextPolish. The resulting assembly consists of 318 contigs and has a total length of 691.96 Mb (Table 2).

Overview of the genome assembly for darkbarbel catfish. (a) Flow chart illustrating the genome assembly pipeline for darkbarbel catfish. (b) Hi-C heatmap (200-kb resolution) showcasing the interaction frequencies between different chromosomes of darkbarbel catfish.
Hi-C scaffolding
The raw Hi-C reads were processed to remove adapters and low-quality bases using Fastp v0.20.111 with parameters -q 20 -l 50. The processed reads were then aligned to the assembly using the Juicer pipeline12. Then the 3D-DNA pipeline13 was used to group the contigs into chromosomes, orient and order the contigs within each chromosome. To further improve the quality of the assembly, we manually corrected the errors using the Juicebox Assembly Tools12. Following the scaffolding procedure, 691.13 Mb were successfully anchored to the 26 chromosomes (Fig. 1b), encompassing an impressive 99.9% of the total assembly size of 692.10 Mb (Table 2). The observed chromosome number concurs with the karyotype analysis reported in the previous study14. The scaffold N50 reached a substantial 27.55 Mb for the final assembly (Table 2). Notably, among the 26 chromosomes, 16 of them exhibited exceptional contiguity with no more than 10 gaps observed (Table 3).
Repeat annotation
RepeatModeler v2.0.215 firstly identified repetitive sequences in the genome assembly using several tools, including RECON, RepeatScout, TRF, Ltr_retriever, and LTRharvest. The identified sequences were then clustered and classified into families using RepeatModeler. The classified libraries were combined with the Teleostei library from Repbase16. RepeatMasker v4.1.417 was performed to mask repetitive sequences in the genome assembly using the combined library generated by RepeatModeler. A significant portion of the genome, approximately 35.79%, is masked, resulting in 247,692,317 bp being identified as repetitive elements. Retroelements, including long terminal repeats (LTRs, 6.90%), long interspersed nuclear elements (LINEs, 5.87%), and short interspersed nuclear elements (SINEs, 1.01%), collectively comprise the largest proportion, occupying 13.77% of the genome (Fig. 2). Furthermore, DNA transposons occupy a notable 11.48% of the genome, while unclassified elements add a nuanced layer, constituting 4.69% of the genomic landscape (Fig. 2).

Genomic landscape of darkbarbel catfish. Circos plot of darkbarbel catfish illustrating from outside to inside, gene density (a), GC content (b), and the densities of DNA transposons (c), LTRs (d), LINEs (e), and SINEs (f), all represented in 200-kb genomic windows.
Gene prediction and function assignment
In this research, we employed a comprehensive approach combining transcriptome-based, de novo, and homology-based methods to predict genes within the genome. For transcriptome-based prediction, RNA-seq reads underwent stringent quality filtering using Fastp v0.20.1, with specific parameters set at -q 20 -l 50. These filtered reads were then aligned to the genome assembly using HISAT2 v2.2.118, followed by assembly using StringTie v2.2.119. Gene structures were subsequently predicted utilizing TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder). For de novo prediction, RNA-seq aligned BAM files served as input for training the AUGUSTUS v3.4.020 gene prediction tool via BRAKER21. This trained model was then employed to predict gene structures within the genome. In the homology-based prediction, we utilized miniport v0.1122 to align protein sequences from P. fulvidraco7, Silurus meridionalis23, and Ictalurus punctatus24 to the genome assembly, enabling the prediction of gene structures based on homologous evidence. To consolidate the results from these three methods, EvidenceModeler25 was employed, enabling the merging and integration of gene predictions. Following the gene prediction, the finalized gene sets derived from preceding methods underwent functional annotation through matching with a variety of databases. In particular, we utilized BLASTP v2.9.026 to align the anticipated genes with SwissProt27, TrEMBL28, eggNOG29, and the NCBI non-redundant (NR) protein databases.
In total, we successfully predicted 22,109 protein-coding genes within the genome (Fig. 2). These predicted genes displayed an average coding sequence length of 1,695.04 bp, an average gene length of ~15 kb, and an average exon number of 10. Further, 96.1% of the total predicted genes, which equates to 21,243 genes, were successfully assigned with at least one functional annotation (Table 4 and Fig. 3).

Venn diagram of function annotations from various databases. The Venn diagram displays the overlap and uniqueness of functional gene annotations derived from 4 databases: TrEMBL, NR, Siwss-Prot, and eggNOG.
Genome synteny analysis
To compare the whole genome synteny, two chromosome-level genomes of Bagridae including yellow catfish30 and Chinese longsnout catfish (Leiocassis longirostris)31 were aligned to the genome assembly of darkbarbel catfish using LAST v135432 with default parameters. The synteny were visualized using Circos v0.69.933. A high degree of synteny conservation between the compared genomes was observed (Fig. 4).

Chromosome sequence synteny comparisons. (a) Syntenic relationship between the darkbarbel catfish genome and the Chinese longsnout catfish genome. (b) Syntenic relationship between the darkbarbel catfish genome and the yellow catfish genome. Each line connects a pair of homologous sequences between the two species.
Data Records
All the raw sequencing data utilized in this study, including WGS, RNA-Seq, and Hi-C, have been deposited in the NCBI database under the BioProject accession number PRJNA819563. Specifically, the Illumina WGS data was archived with the accession number SRR2492634334, while the PacBio WGS data was deposited with the accession number SRR2235495735. The RNA-Seq and Hi-C data sets were archived under the accession numbers SRR2492826336 and SRR2179906337, respectively. The genome assembly is available for public access at the NCBI GenBank under the accession number GCA_030014155.138. Genome annotations, along with predicted coding sequences and protein sequences, can be accessed through the Figshare39.
Technical Validation
To evaluate the completeness of the genome assembly, we used the BUSCO v5.4.240 with the Actinopterygii database (actinopterygii_odb10) to assess the presences of conserved sing-copy genes in the assembly. Out of the total 3,640 BUSCO groups searched, an impressive 99.07% were identified as complete, indicating a high level of gene content preservation. Among these, 98.54% were both complete and present as single-copy genes, further emphasizing the quality of the assembly. Additionally, only 0.33% of the BUSCOs were fragmented, and 0.60% were missing from the assembly (Table 5). This demonstrates the remarkable completeness and conservation of gene content in the darkbarbel catfish genome assembly, achieving one of the best BUSCO scores observed among reported catfish genomes.
To ensure the quality and accuracy of the genome assembly, we employed a two-step validation process. Firstly, the assembly’s Quality Value (QV) was quantified using Merqury41, resulting in a QV score of 40.89, reflecting a high-grade assembly. Then, we mapped the raw sequencing data back to the assembly. For WGS short reads, we utilized BWA v0.7.1742, which resulted in a high mapping rate of 99.79%. For mapping RNA-Seq reads, we used HISAT2 v2.2.1 and achieved an overall mapping rate of 96.44%.
Code availability
No custom software codes were developed as part of this research. All bioinformatics tools and pipelines were executed following the manual and protocols provided by the respective software developers. The versions of the software used, along with their corresponding parameters, have been thoroughly described in the Methods section.
References
Fricke, R., Eschmeyer, W. N. & Fong, J. D. Eschmeyer’s Catalog of Fishes: Genera/Species by Family/Subfamily. https://research.calacademy.org/research/ichthyology/catalog/SpeciesByFamily.asp. Accessed 15 February 2023.
Naylor, R. L. et al. A 20-year retrospective review of global aquaculture. Nature 591, 551–563 (2021).
Tacon, A. G. Trends in global aquaculture and aquafeed production: 2000–2017. Rev. Fish. Sci. Aquac. 28, 43–56 (2020).
Huang, P. et al. Genome-wide association study reveals the genetic basis of growth trait in yellow catfish with sexual size dimorphism. Genomics 114, 110380 (2022).
Liu, Y. et al. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 111, 1258–1265 (2019).
Zhang, G. et al. The effects of water temperature and stocking density on survival, feeding and growth of the juveniles of the hybrid yellow catfish from Pelteobagrus fulvidraco (♀)× Pelteobagrus vachelli (♂). Aquac. Res. 47, 2844–2850 (2016).
Gong, G. et al. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. Gigascience 7, giy120 (2018).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 17, 155–158 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44, e147–e147 (2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Zhang, J. et al. Comparative analysis of the karyotype and nutritional ingredient for the hybrids of Pelteobagrus fulvidraco (♀)× P. vachelli (♂) and their parental fish. Mar. Fish. 39, 149–161 (2017).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 1–6 (2015).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2016).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Zheng, S. et al. Chromosome-level assembly of southern catfish (Silurus meridionalis) provides insights into visual adaptation to nocturnal and benthic lifestyles. Mol. Ecol. Resour. 21, 1575–1592 (2021).
Liu, Z. et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 7, 11757 (2016).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Bairoch, A. & Boeckmann, B. The SWISS-PROT protein sequence data bank: current status. Nucleic Acids Res. 22, 3578 (1994).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Gong, G. et al. Origin and chromatin remodeling of young X/Y sex chromosomes in catfish with sexual plasticity. Natl. Sci. Rev. 10, nwac239 (2023).
He, W. P. et al. Chromosome-level genome assembly of the Chinese longsnout catfish Leiocassis longirostris. Zool. Res. 42, 417–422 (2021).
Kiełbasa, S. M., Wan, R., Sato, K., Horton, P. & Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR24926343 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22354957 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR24928263 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21799063 (2022).
Gong, G. Genbank https://identifiers.org/insdc.gca:GCA_030014155.1 (2023).
Gong, G. et al. Genome annotations of darkbarbel catfish (Pelteobagrus vachelli). figshare https://doi.org/10.6084/m9.figshare.23512404.v2 (2023).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 1–27 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Acknowledgements
This work was supported by the China Agricultural Research System (CARS-46). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
G.G. and J.M. conceived this study. Y.X. and J.H. collected the samples and performed the experiments; G.G., W.K. and Q.L. performed the research and analyzed the data. G.G. drafted the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gong, G., Ke, W., Liao, Q. et al. A chromosome-level genome assembly of the darkbarbel catfish Pelteobagrus vachelli. Sci Data 10, 598 (2023). https://doi.org/10.1038/s41597-023-02509-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02509-0
This article is cited by
-
Chromosome-level genome assembly and annotation of the yellow grouper, Epinephelus awoara
Scientific Data (2024)
