
Abstract
Water chestnut (Trapa L.) is a floating-leaved aquatic plant with high edible and medicinal value. In this study, we presented chromosome-level genome assemblies of cultivated large-seed species Trapa bicornis and its wild small-seed relative Trapa incisa by using PacBio HiFi long reads and Hi-C technology. The T. bicornis and T. incisa assemblies consisted of 479.90 Mb and 463.97 Mb contigs with N50 values of 13.52 Mb and 13.77 Mb, respectively, and repeat contents of 62.88% and 62.49%, respectively. A total of 33,306 and 33,315 protein-coding genes were predicted in T. bicornis and T. incisa assemblies, respectively. There were 159,232 structural variants affecting more than 11 thousand genes detected between the two genomes. The phylogenetic analysis indicated that the lineage leading to Trapa was diverged from the lineage to Sonneratia approximately 23 million years ago. These two assemblies provide valuable resources for future evolutionary and functional genomic research and molecular breeding of water chestnut.
Similar content being viewed by others

Background & Summary
Trapa L., known as water chestnut or water caltrop, is the only genus of Trapaceae. Although the Angiosperm Phylogeny Group (APG) IV treated Trapaceae belonging to Lythraceae, the term “Trapaceae” is still used by some scholars today due to a handful of morphological differences between the two families1. Trapa plants are annual floating-leaved herbs naturally growing in temperate, subtropical and tropical regions of the Old World, and invasive in Australia and North America2. They reproduce sexually and/or asexually and have a high degree of autogamy3,4. The genus has two diversity centers, i.e. the Yangtze River Basin (central China) and the Amur River- Tumen River Basin (the border between China and Russia)5. Trapa plants have high edible value because of their large starchy seeds, which has a long history of consumption. In China, archaeological studies found that water chestnut was widely eaten during the Neolithic Age (7000-2000 BC) with 21 unearthed sites in the basins of the Yellow River and Yangtze River6. In ancient Europe, inhabitants also gathered water chestnut seeds as part of their diet between 4000 and 1000 BC7. The cultivation of water chestnut can be traced back to the Tang (618–907 AD) and Song (916–1279 AD) dynasties8 in the middle and lower reaches of the Yangtze River. At present, it is an important aquatic crop widely grown in China and India9. Additionally, the tender Trapa seeds, stems and leaves are used as vegetables because of the fresh and sweet taste, whereas their seed pericarps are traditional Chinese medicine because of their bioactive components in the treatment of cancer, inflammation and atherosclerosis10,11,12. Furthermore, Trapa has significant ecological value in improving water quality due to its strong absorption capacity for heavy metals and pollutants13.
A better understanding of species identification, evolutionary relationships and genetic information will greatly facilitate the effective management and sustainable utilization of wild plant resources. However, the classification of Trapa species is still open to debate because of their similar morphology of vegetative organs and the highly variable seeds. Some scholars argued that the genus contained more than 20, 30 or 70 species, while others merged them into one or two polymorphic species14. The quantitative taxonomic studies based on morphological variations showed that Trapa species with similar seed sizes were closely related, and all species were divided into two branches, the large- and small-seed clusters15. This was well supported by the molecular studies based on chloroplast (cp) sequences14,16. The cp genome analysis also showed that both the geographical origin and tubercle morphology of seeds were of great significance for deducing relationship within Trapa14. Cytological studies showed two different chromomeric numbers in Trapa (2n = 2x = 48 and 2n = 4x = 96) and suggested that the tetraploid might be a hybrid of diploids17, which was supported by molecular analyses based on allozymes as well as nuclear and chloroplast DNA sequeences18,19. The existence of the two distinct subgenomes was directly confirmed by the recently published chromosome-level assembly of a tetraploid Trapa natans (AABB) genome8. Furthermore, the resequencing data exhibited that large-seed species contained both diploids (2n = 2x = 48, AA) and tetraploids (2n = 4x = 96, AABB), and the small-seed ones only contained diploids (2n = 2x = 48, BB)8. It is a pity that the genome sequences of representatives of the ‘AA’ and ‘BB’ genomes are not available, though such species are very common in the Trapa genus.
Here, we sequenced the genomes of the typical cultivated species Trapa bicornis Osbeck (AA) and a small-seed species Trapa incisa Sieb. et Zucc. (BB), which would greatly deepen the understanding of Trapa diversity and the origin of tetraploid Trapa. De novo assembly using PacBio high-fidelity (HiFi) long reads generated 479.90 and 463.97 Mb contigs for T. bicornis and T. incisa with N50 values of 13.51 and 13.77 Mb, respectively. After scaffolding by Hi-C reads, 98.0% and 98.1% of the contigs could be successfully anchored into 24 pseudo-chromosomes for each genome, respectively. We predicted 33,306 and 33,315 protein-coding genes in T. bicornis and T. incisa genomes, respectively. Despite good collinearity, there were 159,232 structural variations (SVs) identified between the genomes of T. bicornis and T. incisa, overlapping with more than 11 thousand genes. Divergence time estimation indicated that T. bicornis and T. incisa diverged around 1.51 million years ago. The generation of the two genomes provides baseline information of the diversity of Trapa species, which will eventually facilitate functional genomic analysis and molecular breeding of water chestnut.
Methods
Sample collection and sequencing
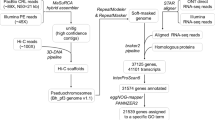
Seeds of T. bicornis and T. incisa were collected from Honghu (29.39°N/113.07°E), Hubei province, China (Fig. 1). Plants were cultured outdoors from March to July in water tanks in Wuhan Botanical Garden, Chinese Academy of Science, Hubei province, China. The 90-day-old individuals for each species were used for the DNA/RNA extractions.

The seeds of T. bicornis Osbeck var. bicornis (a) and T. incisa Sieb. & Zucc. var. incisa (b).
Genomic DNA was isolated from fresh young leaves using Cetyltrimethylammonium bromide (CTAB) method20. A total amount of 1.5 µg DNA per sample was used as input material for the Illumina paired-end library construction. Each library with an average insert size of 350 bp was generated using Truseq Nano DNA HT Sample preparation Kit (Illumina USA) following manufacturer’s instructions. These libraries were sequenced by Illumina HiSeq X Ten system. A total of 125.97 Gb and 53.14 Gb paired-end reads (PE150) covering roughly 183.38 × and 112.42 × of genomes were generated for T. bicornis and T. incisa, respectively (Table 1).
For PacBio long-read sequencing, about 10 µg genomic DNA were sheared into fragments of 10-20 kb in length by g-TUBE (Covaris USA). The fragmented DNA was purified by AMPure PB magnetic beads. The High-fidelity (HiFi) libraries were generated using SMRTbell Express Template Prep Kit 2.0 and sequenced on PacBio Sequel IIe platform (Pacific Biosciences, Menlo Park, USA). A total of 24.11 Gb and 20.42 Gb HiFi reads with N50 sizes of 17,588 bp and 13,963 bp were obtained using the CCS (Circular Consensus Sequencing) software with default parameters (https://ccs.how/), which covered 49.23 × and 43.20 × of T. bicornis and T. incisa genomes, respectively (Table 1).
The high-throughput chromosome conformation capture (Hi-C) libraries were constructed using 5 µg DNA. The DNA crosslinking was performed by 4% formaldehyde. The linked DNA was digested with DpnII restriction endonuclease, labelled with biotin-14-DCTP and then ligated by T4 DNA Ligase. The ligated DNA was sheared into 200-600 bp fragments and sequenced on Illumina HiSeq X Ten system with the paired-end module. About 111.79 Gb and 103.65 Gb of raw data were obtained for T. bicornis and T. incisa, respectively (Table 1).
RNA was extracted from roots, petioles, leaves, flowers and fruits, respectively, using Tiangen RNAprep pure plant kit (Tiangen Biotech, China). Libraries were constructed using NEBNext UltraTM RNA Library Prep Kit (NEB, USA) according to the manufacturer’s instructions, and sequenced on Illumina Novaseq. 6000 platform. RNA-seq datasets from different tissues of the same species were combined as evidence for genome annotation. A total of 34.05 Gb and 36.68 Gb RNA-seq reads were obtained for T. bicornis and T. incisa, respectively (Table 1).
Genome assembly
The PacBio HiFi reads of each genome were de novo assembled by using hifiasm v0.16.121 with default parameters. The assemblies had a total size of 489.65 Mb and 472.74 Mb, containing 325 and 262 contigs with N50 sizes of 13.52 Mb and 13.77 Mb for T. bicornis and T. incisa, respectively (Table 2). The cleaned Hi-C reads were mapped to the corresponding contigs using Juicer v1.9.922. The unique mapped reads were taken as input for 3D-DNA pipeline v18011423 with parameters “-r 0” and then sorted and corrected manually by using JuicerBox v1.11.0824. Finally, a total of 24 pseudo-chromosomes was obtained, which contained 98.01% and 98.14% of the assembled contigs for T. bicornis and T. incisa, respectively (Fig. 2).

Hi-C interactions among the 24 pseudo-chromosomes of T. bicornis (a) and T. incisa (b) genomes. Weak to strong interactions are shown in yellow to red.
We assessed the integrity of the genomes using the BUSCO v5.0 (Benchmarking Universal Single-Copy Orthologs)25 with the ‘embryophyta_odb10’ database. The T. bicornis and T. incisa assemblies contained 97.70% [S:85.10%, D:12.60%, F:0.90%, M:1.40%, n:1614] and 97.80% [S:84.70%, D:13.10%, F:0.80%, M:1.40%, n:1614] of the 1,614 conserved genes, respectively, which are similar to the corresponding values of the diploid T. natans (C: 96.41% [S: 84.76%, D: 11.65%, F: 0.43%, M: 3.16%, n: 1614])26. Based on the Illumina PE150 reads, we assessed the consensus quality values (QV) of the two assemblies using Merqury v2020-01-2927 with “k-mer = 20”. For T. bicornis and T. incisa assemblies, the mapping rate of the reads were 99.88% and 99.61%, respectively, and the QV values were 49.70 and 43.91, respectively (Table 2). These evaluations indicated that the two genome assemblies were of considerable completeness, contiguity and accuracy.
Genome annotation
Custom repeat libraries for each genome were constructed by screening the genome using LTR_finder28, ltrharvest29 and RepeatModeler-2.0.2a30. Then, the non-redundant repeats from Repbase31 and Dfam32 databases were extracted and added to the custom libraries. RepeatMasker v 4.1.2-p1 (http://www.repeatmasker.org) was used to identify repeat sequences based on the custom libraries. A total of 307.95 Mb (62.88%) and 295.42 Mb (62.49%) repetitive sequences were annotated in the T. bicornis and T. incisa genomes, respectively (Table 3).
For protein-coding gene annotation, we employed RNA-seq-based, ab initio and homologue-based predictions to identify gene models. The clean RNA-seq reads were aligned to the assemblies using HISAT2 v2.2.133, and then the alignment was converted to gtf format by StringTie2 v2.1.634. Furthermore, TransDecoder v5.5.035 was used to identify the open reading frame (ORF) and modify the boundaries of exons. The ab initio gene predictions were generated by three de novo predicting programs, including Augustus-3.3.336, SNAP v2006-07-2837 and GlimmerHMM 3.0.438,39. Proteins from Punica granatum40, Arabidopsis thaliana TAIR1041, Eucalyptus grandis42, Melaleuca alternifolia43 and tetraploid Trapa natans8 were aligned to the genomes using TBLASTN44. The homologous genes were identified using Exonerate v2.2.045. The RNA-seq evidences, ab initio predictions and homolog evidences were fed to MAKER v3.0146 to generate the final gene set. A total of 33,306 and 33,315 protein-coding genes were predicted in the T. bicornis and T. incisa genomes, respectively.
Functional annotation of protein-coding genes were evaluated based on five public databases, including GO (http://geneontology.org/), KEGG (https://www.kegg.jp/), GenBank nr (https://www.ncbi.nlm.nih.gov/), Uniprot (https://www.uniprot.org/) and Interpro (http://www.ebi.ac.uk/interpro/), using DIAMOND v2.0.13.15147. A total of 31,360 (94.14%) and 31,406 (94.27%) genes were successfully annotated in at least one database for T. bicornis and T. incisa, respectively (Table 3). The BUSCO completeness values were 97.70% and 98.10% of the predicted proteins of T. bicornis and T. incisa, respectively (Table 3).
Variations between the T. bicornis and T. incisa genomes
Single nucleotide polymorphisms (SNPs) between the genomes of T. bicornis and T. incisa were detected by alignment of the two assemblies using NUCmer from MUMMER448. We set the minimum alignment length to 100 bp and retained the uniquely matching fragments. A total of 9,449,234 SNPs were identified by show-snps tool from MUMMER448 (Fig. 3).

Genomic landscape of T. bicornis and T. incisa. Window size is 500 kb. The cycles from outer to inner show (I) densities of repetitive sequences, (II) gene, (III) SNP and (IV) SV numbers in sliding windows. All statistics were normalized by log scale.
To identify SVs, T. incisa genome was mapped to T. bicornis genome by using Minimap249 with the parameter “-ax asm5”. Assemblytics was adopted to extract unique alignments and identify SVs based on them50. Protein-coding genes overlapping with SV regions were retrieved by BEDTools v2.29.151. The final SVs were classified into seven categories: deletion, insertion, repeat contraction, repeat expansion, tandem contraction, tandem expansion and substitution. A total of 159,232 SVs were identified between T. bicornis and T. incisa genomes, which accounted for 110.49 Mb and 140.13 Mb sequences of the two genomes, respectively (Table 4). These SVs overlapped with 11,265 and 11,621 genes of the two Trapa genomes, respectively.
The synteny between the published tetraploid T. natans genome and the present two diploid Trapa genomes
Our new assemblies provided great resource for investigating the origin of the Trapa tetraploid and the genomic changes post-polyploidization. The genomes of T. bicornis and T. incisa and the two subgenomes of the published tetraploid genome were pairwise aligned with each other by using MUMMER448 (Fig. 4). The syntenic regions were extracted from the alignments with the software syri-1.452. Clearly, the T. bicornis and T. incisa genomes possessed the highest percentage of syntenic regions with the A and B subgenomes of T. natans, respectively, suggesting that the formers represented the ancestry genomes of the latter two, separately. The percentage of syntenic regions between the A and B subgenomes (69.01%) was higher than that between the T. bicornis and T. incisa genomes (59.81%), evidencing homoeologous recombination events after tetraploidization53.

Synteny between genomes of T. bicornis, T. incisa and subgenomes of tetraploid T. natans. (a) Pairwise comparisons of the genomes of T. bicornis, T. incisa and the two subgenomes of tetraploid T. natans. (b) The percentages of syntenic regions of each comparison.
Comparative genomics and divergence time estimation
Using OrthoFinder v2.5.254, orthologous groups were constructed for 11 species, including Arabidopsis thaliana41, Brassica oleracea55, Citrus sinensis56, Corymbia citriodora26, Eucalyptus grandis42, Melaleuca alternifolia43, Punica granatum40, Sonneratia alba57, Trapa bicornis, Trapa incisa and tetraploid Trapa natans8 (AABB), which was divided into two subgenomes. A total of 1,105 single copy orthologues were obtained, and they were aligned using MUSCLE v3.8.3158. The alignments of protein sequence were converted into nucleotide sequences. The final alignments of orthologous groups were concatenated to build a maximum likelihood phylogenetic tree using RAxML-8.2.1259 with “GTRGAMMA” model. The figure of phylogenetic tree was visualized by iTOLv660. Divergence times among the species were estimated using the MCMC tree program implemented in PAML v4.9i61. The reference divergence time was obtained from http://timetree.org/. The three species (Citrus sinensis, Arabidopsis thaliana and Brassica oleracea) were constrained as root in the time-calibrated phylogeny. Due to the lack of strong morphological evidence, the relationship between Trapa and Lythraceae has been unclear historically62. Here, our phylogenetic tree (Fig. 5) showed that Trapa was sister to the genus Sonneratia (Lythraceae s.l.), which was also supported by previous studies based on chloroplast and nuclear sequences14,63,64. According to the time-calibrated phylogeny, the Trapa-Sonneratia clade diverged from Punica (Lythraceae) at ca 35.24 million years ago. Then, the two genera (Trapa and Sonneratia) diverged ca 23 Mya ago, and the two Trapa species with distinct genomes (T. bicornis: AA; T. incisa: BB) diverged ca 1.5 Mya.

Phylogenetic tree with estimated divergence times. The maximum likelihood tree was constructed based on 1,106 single-copy orthologous genes. The red dots at the nodes indicated that the values were supported by fossil evidence.
Data Records
The raw data of Illumina PE150 reads, PacBio HiFi long reads and Hi-C reads from T. bicornis were submitted to the National Center for Biotechnology Information (NCBI) SRA (Sequence Read Archive) database with accession number SRR2218506865, SRR2218506766, SRR2218506667 under BioProject accession number PRJNA89343168. The RNA-seq data for the five tissues are also under the PRJNA89343168. For T. incisa, the raw data of Illumina, PacBio and Hi-C sequencing had been deposited in SRA database as SRR2209461469, SRR2209461370 and SRR2209461271 under PRJNA89409472. And the RNA-seq data are also under the same BioProject accession. The assembly genome files were stored in GenBank database under the accession GCA_030064425.173 and GCA_030064435.174, respectively. The genomes and annotation files and raw sequencing data have also been uploaded in National Genomics Data Center (NGDC) under PRJCA01213375 and PRJCA01213476.
Technical Validation
The quality scores across all bases and GC content of the Illumina raw sequencing data were inspected by FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Contig level and chromosome level of the assemblies were assessed in four ways: N50 for continuity, QV for accuracy, BUSCO for completeness and paired-end reads mapping rate for consistency with raw data. The protein-coding genes were verified by values of BUSCO and functional databases annotation. For construction of phylogenetic tree, each branch received 100% bootstrap values.
Code availability
The scripts and command lines were uploaded on the github (https://github.com/fcbayern31/A-pipeline-for-common-genomic-analysis.git). All softwares, which are in the public domain, were used in accordance with the official instructions. Anything not specified in the method is executed with default parameters.
References
The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016).
Chen, J., Ding, B. Y. & Funston, M. Trapaceae. In Flora of China 13, 290–291 (2007).
Arima, S., Daigoho, M. & Hoque, M. A. Flower development and anthesis behavior in the water chestnut (Trapa sp.). Bull. Fac. Agric. 84, 83–92 (1999).
Li, X., Fan, X., Chu, H., Li, W. & Chen, Y. Genetic delimitation and population structure of three Trapa taxa from the Yangtze River, China. Aquat. Bot. 136, 61–70 (2017).
Xue, Z., Xue, J., Victorovna, K. & Ma, K. The complete chloroplast DNA sequence of Trapa maximowiczii Korsh. (Trapaceae), and comparative analysis with other Myrtales species. Aquat. Bot. 143, 54–62 (2017).
Guo, Y., Wu, R., Sun, G., Zheng, Y. & Fuller, B. T. Neolithic cultivation of water chestnuts (Trapa L.) at Tianluoshan (7000-6300 cal BP), Zhejiang Province, China. Sci. Rep. 7, 16206 (2017).
Karg, S. The water chestnut (Trapa natans L.) as a food resource during the 4th to 1st millennia BC at Lake Federsee, Bad Buchau (southern Germany). Environ. Archaeol. 11, 125–130 (2006).
Lu, R. et al. Genome sequencing and transcriptome analyses provide insights into the origin and domestication of water caltrop (Trapa spp., Lythraceae). Plant Biotechnol. J. 20, 761–776 (2022).
Hummel, M. & Kiviat, E. Review of world literature on water chestnut with implications for management in North America. J. Aquat. Plant Manage. 42, 17–28 (2004).
Ciou, J., Wang, C., Chen, J. & Chiang, P. Total phenolics content and antioxidant activity of extracts from dried water caltrop (Trapa taiwanensis nakai) hulls. J. Food Drug Anal. 16, 41–47 (2008).
Yu, H. & Shen, S. Phenolic composition, antioxidant, antimicrobial and antiproliferative activities of water caltrop pericarps extract. Lwt-Food Sci. Technol. 61, 238–243 (2015).
Kauser, A. et al. In vitro antioxidant and cytotoxic potential of methanolic extracts of selected indigenous medicinal plants. Prog. Nutr. 20, 706–712 (2018).
Xu, L. et al. Assessment of the nutrient removal potential of floating native and exotic aquatic macrophytes cultured in swine manure wastewater. Int. J. Environ. Res. Public Health 17, 1103 (2020).
Fan, X. et al. Fifteen complete chloroplast genomes of Trapa species (Trapaceae): insight into genome structure, comparative analysis and phylogenetic relationships. BMC Plant Biol. 22, 230 (2022).
Fan, X. et al. Analysis of morphological plasticity of Trapa L. from China and their taxonomic significance. Plant Sci. J. 34, 340–351 (2016).
Wang, W., Fan, X., Li, X. & Chen, Y. The complete chloroplast genome sequence of Trapa incisa Sieb. & Zucc. (Lythraceae). Mitochondrial DNA B Resour. 6, 1732–1733 (2021).
Oginuma, K., Takano, A. & Kadono, Y. Karyomorphology of some Trapaceae in Japan. Acta Phytotax. Geobot. 47, 47–52 (1996).
Kim, C., Ryun, N. H. & Choi, H. Molecular genotyping of Trapa bispinosa and T. japonica (Trapaceae) based on nuclear AP2 and chloroplast DNA trnL-F region. Am. J. Bot. 97, e149–152 (2010).
Takano, A. & Kadono, Y. Allozyme variations and classification of Trapa (Trapaceae) in Japan. Aquat. Bot. 83, 108–118 (2005).
Doyle, J. & Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Cheng, H., Concepcion, G., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6, 256–258 e251 (2018).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
He, Z. et al. Evolution of coastal forests based on a full set of mangrove genomes. Nat. Ecol. Evol 6, 728–749 (2022).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110, 462–467 (2005).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 41, D70–82 (2013).
Kim, D., Paggi, J., Park, C., Bennett, C. & Salzberg, S. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Majoros, W. H. & Salzberg, S. L. An empirical analysis of training protocols for probabilistic gene finders. BMC Bioinformatics 5, 206 (2004).
Luo, X. et al. The pomegranate (Punica granatum L.) draft genome dissects genetic divergence between soft- and hard-seeded cultivars. Plant Biotechnol. J. 18, 955–968 (2020).
Berardini, T. Z. et al. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53, 474–485 (2015).
Myburg, A. A. et al. The genome of Eucalyptus grandis. Nature 510, 356–362 (2014).
Voelker, J., Shepherd, M. & Mauleon, R. A high-quality draft genome for Melaleuca alternifolia (tea tree): a new platform for evolutionary genomics of myrtaceous terpene-rich species. Gigabyte 2021, 1–15 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Marcais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Nattestad, M. & Schatz, M. C. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023 (2016).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Goel, M. et al. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome biol. 20, 1–13 (2019).
Gaeta, R. T. & Chris, P. J. Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytologist 186, 18–28 (2010).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Parkin, I. A. et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, R77 (2014).
Wang, L. et al. Somatic variations led to the selection of acidic and acidless orange cultivars. Nat. Plants 7, 954–965 (2021).
Healey, A. L. et al. Pests, diseases, and aridity have shaped the genome of Corymbia citriodora. Commun. Biol. 4, 537 (2021).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49, W293–W296 (2021).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Graham, S. A., Crisci, J. V. & Hoch, P. C. Cladistic analysis of the Lythraceae sensu lato based on morphological characters. Bot. J. Linn. Soc. 113, 1–33 (1993).
Graham, S. A., Hall, J., Sytsma, K. & Shi, S. H. Phylogenetic analysis of the Lythraceae based on four gene regions and morphology. Int. J. Plant Sci. 166, 995–1017 (2005).
Huang, Y. L. & Shi, S. H. Phylogenetics of Lythraceae sensu lato: a preliminary analysis based on chloroplast rbcL gene, psaA-ycf3 spacer, and nuclear rDNA internal transcribed spacer (ITS) sequences. Int. J. Plant Sci. 163, 215–225 (2002).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR22185068 (2022).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR22185067 (2022).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR22185066 (2022).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA893431 (2022).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR22094614 (2022).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR22094613 (2022).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR22094612 (2022).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA894094 (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/assembly/GCA_030064425.1 (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/assembly/GCA_030064435.1 (2022).
NGDC BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA012133 (2022).
NGDC BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA012134 (2022).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (32170395 and 82060684), the Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Talent Program of Wuhan Botanical Garden, Chinese Academy of Sciences (Y855291), Young and Middle-Aged Talents Training Program of Traditional Chinese Medicine of Jiangxi Province (2020-2) and Jiangxi University of Chinese Medicine Science and Technology Innovation Team Development Program (CXTD22002).
Author information
Authors and Affiliations
Contributions
L.G. and Y.C. conceived this project; X.F. and Y.C. collected the samples; M.Q. and C.H. performed the data analyses; M.Q. and Y.C. wrote the manuscript; L.G., Y.C., Y.X., Y.Z., S.W., W.L. and S.G. revised the manuscript. All authors have read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qu, M., Fan, X., Hao, C. et al. Chromosome-level assemblies of cultivated water chestnut Trapa bicornis and its wild relative Trapa incisa. Sci Data 10, 407 (2023). https://doi.org/10.1038/s41597-023-02270-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02270-4
