
Abstract
X-ray absorption spectroscopy (XAS) is a premier technique for materials characterization, providing key information about the local chemical environment of the absorber atom. In this work, we develop a database of sulfur K-edge XAS spectra of crystalline and amorphous lithium thiophosphate materials based on the atomic structures reported in Chem. Mater., 34, 6702 (2022). The XAS database is based on simulations using the excited electron and core-hole pseudopotential approach implemented in the Vienna Ab initio Simulation Package. Our database contains 2681 S K-edge XAS spectra for 66 crystalline and glassy structure models, making it the largest collection of first-principles computational XAS spectra for glass/ceramic lithium thiophosphates to date. This database can be used to correlate S spectral features with distinct S species based on their local coordination and short-range ordering in sulfide-based solid electrolytes. The data is openly distributed via the Materials Cloud, allowing researchers to access it for free and use it for further analysis, such as spectral fingerprinting, matching with experiments, and developing machine learning models.
Similar content being viewed by others
Background & Summary
The glass/ceramic lithium thiophosphates (gc-LPS) along the composition line Li2S–P2S5 are considered promising electrolytes for solid-state batteries because of their superionic lithium conductivity at room temperature (>10–3 Scm–1), soft mechanical properties, and low grain boundary resistance1,2. Although gc-LPS lacks long-range atomic ordering, it exhibits characteristic short-ranged structural motifs that vary with the LPS composition and can affect the Li conductivity.
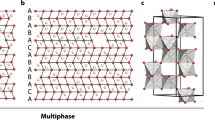
Figure 1 illustrates how the local coordination of S atoms with Li and P atoms in the crystalline phases of LPS changes with increasing Li2S content x in (Li2S)x(P2S5)1–x: P2–S and P–S–Li2 in LiPS3 ((Li2S)0.5(P2S5)0.5); P2–S–Li, P–S–Li2, P–S–Li3 and P–S–Li4 in Li7P3S11 ((Li2S)0.7(P2S5)0.3); P–S–Li2, P–S–Li3 and P–S–Li4 in Li3PS4 ((Li2S)0.75(P2S5)0.25); P–S–Li3, S–Li6 and S–Li7 in Li7PS6 ((Li2S)0.875(P2S5)0.125). To understand the short-range ordering and its impact on Li conductivity in gc-LPS, several characterization tools have previously been employed, including X-ray diffraction (XRD)3,4,5,6,7,8,9,10,11,12, Raman spectroscopy3,10,12,13,14,15,16,17,18, nuclear magnetic resonance (NMR)10,12,15,19,20,21, X-ray photoelectron spectroscopy (XPS)16,22,23,24,25,26,27,28,29,30, and X-ray absorption spectroscopy (XAS)16,24,31,32,33,34.

Schematic illustration of the local coordination of S atoms with Li and P atoms in selected (Li2S)x(P2S5)1–x crystalline structures. Li: green; S: yellow; P: purple. LiPS3: orange region; Li7P3S11: red region; Li3PS4: green region; Li7PS6: blue region.
As Fig. 1 indicates, the average Li coordination number of S increases with the fraction of Li2S in LPS, leading to a greater density of Li around S atoms. Such an increased local Li concentration has previously been argued to increase electron density and thereby give rise to a shielding effect around S atoms and a red shift of the S K-edge24. Tender energy XAS spectroscopy is therefore a natural choice to probe the local geometric and electronic structures in gc-LPS.
Sulfur is known to participate to a greater extent than phosphorus in interfacial reactions during cycling, forming Li2S at the negative electrode or other metal sulfides at the positive electrode (e.g., NiS)33. In contrast, phosphorus is mostly bound in the center of PS43– tetrahedra (as P5+ species), except for the direct P–P bonding in P2S64– motifs (as P4+ species). There is no direct Li–P bonding in gc-LPS and hence sulfur is more sensitive to the change in Li stoichiometry. Based on these considerations, sulfur K-edge spectroscopy can be expected to yield important insights into the electrochemical reactions in LPS-based solid-state batteries.
Commonly, XAS spectra are interpreted by comparison with characteristic features in spectra taken from reference materials, however, this approach is challenging when the composition and structure of the material cannot be readily identified35. In order to aid with the interpretation of XAS measurements and to understand the nature of the short-range ordering and its impact on properties such as Li conductivity and the electronic structure, first-principles XAS simulations have previously been conducted35,36,37,38,39,40,41,42,43. These simulations involve the modelling of the excitation of a core electron into the conduction bands, leaving behind a core hole. Within methods based on density functional theory (DFT) band structure, two approaches are commonly used to account for the core-hole final state effect: (i) the excited electron and core-hole (XCH) method with self-consistent relaxation of valence electrons44,45,46, which is implemented in, e.g., XSPECTRA47 and the Vienna Ab Initio Simulation Package (VASP)48, and (ii) many-body perturbation theory based on the Bethe-Salpeter equation treating the screening of valence electrons with linear response, which is implemented in, e.g., OCEAN49 and EXCITING50. Generally, the many-body perturbation theory-based method is computationally more demanding35. In comparison, the XCH approach can provide a reasonable accuracy that is sufficient to compare trends with experimental measurements at a moderate computing time36,44,51,52,53,54 and is therefore a good choice for the compilation of a large XAS database. In addition, Pascal et al. demonstrated that the XCH approach can reliably predict the features of the S K-edge of distinct coordination environments in Li-S batteries36.
XAS simulations have so far been limited to crystalline LPS phases, and to our knowledge no XAS simulations of gc-LPS have been reported owing to the complexity of the glassy phases. We recently mapped the phase diagram of gc-LPS by combining DFT, artificial neural network (ANN) potentials, genetic-algorithm (GA) sampling, and ab initio molecular dynamics (AIMD) simulations, to compile a database of stable and metastable gc-LPS atomic structures55. This gc-LPS phase diagram is the foundation for the herein reported database of simulated gc-LPS S K-edge XAS spectra.
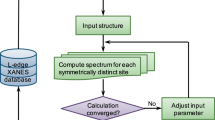
Here, we report the S K-edge XAS simulations for an extensive database of LPS/gc-LPS structures. A workflow for automated calculations using the XCH approach (Fig. 2) was implemented using the open-source Pymatgen package56 and VASP48. The final database contains 2681 simulated S K-edge XAS spectra for 66 crystalline and glassy structures. Where possible, the simulated spectra were benchmarked by comparison with tender energy XAS spectroscopy measurements. The database is distributed via the Materials Cloud repository57 and the workflow available open source, enabling open access by other researchers for further exploration.

Flowchart with the workflow for building the sulfur K-edge XAS spectral database of crystalline and amorphous lithium thiophosphate materials.
Methods
Density functional theory calculations
All DFT calculations were carried out within the projector-augmented-wave (PAW) approach58,59 as implemented in VASP58,60. The simulation parameters were carefully tested to ensure numerical convergence with the energy cut-off for the plane-wave basis set, the supercell size for XCH calculations, the density of the k-point meshes, and the number of unoccupied bands. Different exchange-correlation functionals, pseudopotentials from the VASP library, and core-hole charges (full vs. half) were compared to determine the optimal VASP input parameters for XAS simulations. The following parameters yielded converged results that compared best with experimental reference spectra.
Ground-state energy calculations and XAS simulations were performed with the local-density approximations (LDA) exchange-correlation functional61 and the VASP GW pseudopotentials, which achieve a more accurate description of the post-edge region than regular LDA potentials because the GW pseudopotentials were optimized to yield more accurate scattering properties at high energies well above the Fermi level. A kinetic energy cut-off for the wavefunction of 400 eV, supercells with an edge length of at least 10~15 Å in each direction, and a full core hole were used for all calculations. Three times as many unoccupied bands as occupied bands were included in all calculations to ensure the convergence of the conduction band in the relevant energy range. Gaussian smearing with a width of 0.05 eV was used, and total energies were converged to better than 10−5 eV/atom. The first Brillouin zone was sampled using Γ-centered k-point meshes with the resolution of 0.25 Å−1 generated with VASP’s automatic sampling method. In XCH simulations, a constant Lorentzian broadening of 0.05 eV was introduced. Additional broadening can be added during post-processing when compared to experiment as discussed below.
In XAS simulations with the XCH approach, the final state is treated self-consistently subject to the presence of a core-hole in the S 1 s orbital44,53. The XAS spectrum is calculated as the imaginary part of the frequency-dependent dielectric matrix averaged over the diagonal matrix elements within the PAW frozen-core approximation. For comparison with measured reference spectra, the simulated XAS spectra were convoluted with a Gaussian function with a full width at half maximum of 0.5 eV to simulate instrument broadening and with a Lorentzian function with an energy-dependent width of 0.59 eV + a × (Ec-Ecbm) to simulate the core-hole lifetime broadening and quasiparticle lifetime broadening, where a is a fitting parameter chosen to be 0.1 in this study and Ec and Ecbm are DFT energy levels of conduction bands and conduction band minimum, respectively. Each absorption edge was aligned using the excitation onset determined from the total energy difference between the final state and the initial state, following a previously reported procedure54.
Structure selection
The structures for the XAS simulations were selected from the LPS structure library by Guo et al.55 The gc-LPS structures in this dataset were generated by iterative manipulation of the known crystal structures along the (Li2S)x(P2S5)1–x composition line (LiPS3, Li4P2S7, Li7P3S11, α-Li3PS4, β-Li3PS4, γ-Li3PS4, and Li7PS6) using a previously established protocol62,63. In short, (A) a supercell of a crystal structure was created, (B) either Li and S atoms were removed with a ratio of 2:1 (Li2S), or P and S atoms were removed with a ratio of 2:5 (P2S5), and (C) low-energy configurations of the new composition were determined with a genetic (evolutionary) algorithm using an artificial neural network (ANN) interatomic potential as implemented in the atomic energy network (ænet) package64,65,66. For further details we refer the reader to ref. 55. From this dataset, the (Li2S)x(P2S5)1–x structures with the lowest formation energies relative to Li2S and P2S5 at each composition were chosen for XAS simulations. In addition, the above crystalline LPS compounds and the crystal structures of the sulfur-deficient Li2PS3 and Li48P16S61 were included.
Automated DFT workflow for constructing XAS database
On the basis of the determined parameters from the benchmark systems, a workflow was devised for automated XCH calculations for generating an XAS database (Fig. 2). For each optimized LPS structure, the workflow based on symmetry automatically determines the inequivalent S sites and their respective weights. Our implementation makes use of the symmetry tools from the Pymatgen package56. Pymatgen functions were further used to create supercells and generate VASP input files for single-point LDA calculations to obtain the ground state energy, and for XCH calculations for all symmetrically distinct S atoms in the supercell. Raw data from completed DFT calculations are post-processed, which mainly involves two steps: (1) applying the peak alignment to distinct S atoms using the excitation onset determined from the total energy difference between the final state and the initial state and (2) averaging the aligned spectra with the correct weights to compute the XAS spectrum of the whole system. Note that the data without averaging contains information about the XAS features of local atomic structures, which could be used for further exploration, e.g., machine-learning-assisted spectral interpretation.
Sample preparation and XAS measurements
The experiment S K-edge XAS spectra were measured at the 8-BM and 7-ID-2 beamlines at National Synchrotron Light Source II (NSLS-II). The P2S5 spectrum was measured at 8-BM in fluorescence yield (FY) mode, and Li2S, NiS, and β-LPS were measured at 7-ID-2 in electron yield (EY) mode. We used unfocused beam with spot sizes of 2.5 mm × 5 μm and 1 mm × 1 mm at 8-BM and 7-ID-2, respectively.
Prior to the measurements, the samples were pressed into pellets with 1 cm diameter. For the 8-BM measurement, the sample was sealed between Kapton tape and polypropylene film in an argon-filled glovebox, and then transferred into the helium chamber at the beamline. For the 7-ID-2 measurements, the samples were mounted on a sample bar and sealed in an aluminized polymer bag in the glovebox, and then transferred into the vacuum chamber at the beamline using an argon-filled transfer bag. The experiment XAS spectra were processed with Athena software package67.
Data Records
The database contains 66 structures with between 12 and 162 atoms, 18 of which are crystalline and the rest are amorphous (see Table 1). For each structure, a ground state self-consistent field (SCF) calculation is computed and stored in a directory named input_SCF. For every symmetrically inequivalent S site (between 1 and 86 per structure), a core-hole calculation is performed using the S core-hole pseudopotential. For each individual VASP calculation, we provide all input files except the pseudopotentials (POTCAR files) since those are distributed with VASP: INCAR, POSCAR and KPOINTS. That way, calculations can be rerun after reconstructing the appropriate potential file for each calculation. Due to the large size of many VASP output files, we only keep those necessary for presenting and reproducing the spectral database. These include the INCAR, POSCAR and KPOINTS input files, and the OSZICAR output file (to demonstrate the convergence of the calculation). Additionally, we save the Fermi energy where relevant (efermi.txt) and post-process the XAS from the OUTCAR (mu.dat; note: most regions with zero intensity are discarded to save space). Each spectrum consists of four columns: the energy, and the three components of the XAS (corresponding to the three polarization directions along the Cartesian coordinates). VASP 6.2.1 was used with GPU acceleration, and no post-processing was performed, such that the database is essentially preserved exactly as output by VASP. We provide short post-processing scripts for extracting key observables, such as the energy and spectral intensity, as well as a supplementary tarball with the site-averaged spectra for each material. The spectral data are stored in the Materials Cloud (https://www.materialscloud.org)68.
Technical Validation
Benchmark of the XAS simulations
Our calculations started with the benchmark of the XAS simulations using reference sulfur compounds. Some of the most relevant compounds from gc-LPS/electrode interfacial degradation, including Li2S, P2S5 and NiS, were selected as validation systems. To validate the VASP XAS simulations, the simulated spectra were compared against experimental measurements for three benchmark systems (Li2S, P2S5 and NiS) as shown in Fig. 3. The simulated spectra successfully reproduce the main features in reference systems. It is known that Kohn-Sham DFT underestimates band gaps and concomitantly band widths, due to inaccurate estimates of quasiparticle (excitation) energies based solely on the Kohn-Sham eigenspectrum36. Therefore, the calculated XAS spectra may underestimate peak separations compared to experiments, as seen in the Li2S spectrum.

Benchmark results of XAS simulations of (a) Li2S, (b) P2S5, (c) NiS, and (d) β-Li3PS4. In each subfigure, the black curve indicates the experimental spectrum, and the red curve indicates the simulated spectrum. Spectra calculated for different distinct S sites are shown as dashed lines in (b,d). The experimental spectra in (a,c,d) were measured in electron yield mode at Beamline 7-ID-2 of NSLS-II. The experimental spectrum in (b) was measured in fluorescence yield (FY) mode at Beamline 8-BM of NSLS-II, with the green solid line showing the original FY data, and black solid line showing the self-absorption-corrected data. DFT calculations with (orange) and without Hubbard U correction for Ni (red) are shown in (c). The labels of different S sites in (d) correspond to the local motifs in Fig. 1b.
The XCH simulations successfully reproduce the three main features in the Li2S spectrum at 2474, 2476.5, and 2484 eV, as well as the peak shoulder at 2480 eV, while the energy separation of the first two peaks was slightly underestimated. The relative intensities of these peaks are mostly successfully reproduced, with a slight underestimate of the intensity of the third peak at 2484 eV.
The spectrum of P2S5 in the main edge region between 2470 to 2474 eV exhibits a two-peak feature, originating from S sites under two different local chemical environments. Note that the experimental spectrum of P2S5 in Fig. 3b was measured in fluorescence yield (FY) mode and is plotted with a green solid line, where the peaks are damped due to self-absorption (SA) effect. To better compare the experimental spectrum with the simulated data, we performed a SA correction using Athena software67. The structure of (P2S5)2 is composed of two types of S atoms: 4 terminal S and 6 bridging S, denoted as S(1) and S(2), respectively in Fig. 3b and indicated in structural motifs in Fig. 1. The terminal S atom is coordinated with one P atom, and the P-S bond length is around 1.9 Å. In comparison, the bridging S atom is coordinated with two P atoms; the charge distribution over the bridging S is less negative than the terminal S, leading to a longer P-S bond length of 2.1 Å and blueshift of the main peak as shown in Fig. 3b. Our results are the first demonstration that the main edge of the S K-edge in P2S5 can be attributed to two types of S atoms with different local coordinations. This also demonstrates that XCH simulations can distinguish the inequivalent absorption sites.
To study the interfacial reaction between LPS and Ni-based cathodes, we also computed S K-edge XAS for NiS, a common degradation product of LPS in contact with Ni-based cathode materials, without and with a Hubbard U correction of 3.9 eV to account for the correlation of the Ni d-band electrons. As shown in Fig. 3c, the Hubbard U correction does not change the main absorption edge, but leads to a broadened absorption edge and increased intensity in the post-edge region. From the DFT + U simulation, there are three peaks in the 2475–2485 eV region at 2476, 2477.6, and 2480.5 eV. From the experiment, those peaks are at 2476, 2478.3, and 2483 eV. The peak separation is underestimated in the simulation, likely due to inaccurate DFT energy levels, similar to the issues we have seen in Fig. 3a. While the U value is dependent on the species and materials and must be tested carefully, the overall sufficiently good agreement between the XCH simulations and experiments on exemplary reference compounds demonstrates the robustness of our approach and the reliability of our dataset41.
Validation of DFT calculated S K-edge in crystalline β-Li3PS4
To further validate the simulated XAS spectra of the sampled gc-LPS phases, we also conducted experimental measurements of XAS reference data for LPS crystal structures. The S K-edge in crystalline β-Li3PS4 was measured under fluorescence mode at NSLS-II. As shown in Fig. 3d, our computed XAS spectra are in good agreement with the experimental data. The absorption edge is around 2471 eV, which is likely due to the S 1 s to S 3p σ* transition (dumbbell-shaped S22–)24. The simulated XAS not only reproduces most features, but also yields a comparable peak splitting for the S K-edge in the β-Li3PS4 crystal. In β-Li3PS4, there are three inequivalent S sites (denoted as S(3), S(4), S(5) in Figs. 1, 3d), where the local coordination is shown in Fig. 1. While the charge distribution at the three S sites is comparable, the P–S and Li–S bond lengths exhibit a sizable variation. In this case, the core-level chemical shift cannot be simply explained by the bond length and charge transfer. It is an interesting future research direction to develop optimal structural and chemical descriptors for the interpretation of XAS spectral features in gc-LPS and especially the interphases at solid-state interfaces with Li metal anodes and Ni-based cathodes.
Usage Notes
See Code availability.
Code availability
Short scripts used for extracting useful information from the VASP output files, such as the XAS and energies, are provided with the database. The workflow is available on GitHub (https://github.com/atomisticnet/xas-tools/releases/tag/v0.1.0).
References
Kudu, Ö. U. et al. A Review of Structural Properties and Synthesis Methods of Solid Electrolyte Materials in the Li2S − P2S5 Binary System. J. Power Sources 407, 31–43 (2018).
Kim, K. J., Balaish, M., Wadaguchi, M., Kong, L. & Rupp, J. L. M. Solid-State Li–Metal Batteries: Challenges and Horizons of Oxide and Sulfide Solid Electrolytes and Their Interfaces. Adv. Energy Mater. 11, 2002689 (2021).
Mercier, R., Malugani, J. P., Fahys, B., Douglande, J. & Robert, G. Synthese, Structure Cristalline et Analyse Vibrationnelle de l’hexathiohypodiphosphate de Lithium Li4P2S6. J. Solid State Chem. 43, 151–162 (1982).
Murayama, M., Sonoyama, N., Yamada, A. & Kanno, R. Material Design of New Lithium Ionic Conductor, Thio-LISICON, in the Li2S–P2S5 System. Solid State Ionics 170, 173–180 (2004).
Yamane, H. et al. Crystal Structure of a Superionic Conductor, Li7P3S11. Solid State Ion. 178, 1163–1167 (2007).
Homma, K., Yonemura, M., Nagao, M., Hirayama, M. & Kanno, R. Crystal Structure of High-Temperature Phase of Lithium Ionic Conductor, Li3PS4. J. Phys. Soc. Jpn. 79, 90–93 (2010).
Onodera, Y. et al. Crystal Structure of Li7P3S11 Studied by Neutron and Synchrotron X-ray Powder Diffraction. J. Phys. Soc. Jpn. 79, 87–89 (2010).
Homma, K. et al. Crystal Structure and Phase Transitions of the Lithium Ionic Conductor Li3PS4. Solid State Ion. 182, 53–58 (2011).
Onodera, Y., Mori, K., Otomo, T., Sugiyama, M. & Fukunaga, T. Structural Evidence for High Ionic Conductivity of Li7P3S11 Metastable Crystal. J. Phys. Soc. Jpn. 81, 044802 (2012).
Dietrich, C. et al. Local Structural Investigations, Defect Formation, and Ionic Conductivity of the Lithium Ionic Conductor Li4P2S6. Chem. Mater. 28, 8764–8773 (2016).
Hood, Z. D. et al. Structural and Electrolyte Properties of Li4P2S6. Solid State Ion. 284, 61–70 (2016).
Dietrich, C. et al. Synthesis, Structural Characterization, and Lithium Ion Conductivity of the Lithium Thiophosphate Li2P2S6. Inorg. Chem. 56, 6681–6687 (2017).
Busche, M. R. et al. In Situ Monitoring of Fast Li-Ion Conductor Li7P3S11 Crystallization Inside a Hot-Press Setup. Chem. Mater. 28, 6152–6165 (2016).
Ohara, K. et al. Structural and Electronic Features of Binary Li2S-P2S5 Glasses. Sci. Rep. 6, 21302 (2016).
Dietrich, C. et al. Lithium Ion Conductivity in Li2S–P2S5 Glasses – Building Units and Local Structure Evolution During the Crystallization of Superionic Conductors Li3PS4, Li7P3S11 and Li4P2S. J. Mater. Chem. A 5, 18111–18119 (2017).
Hakari, T. et al. Structural and Electronic-State Changes of a Sulfide Solid Electrolyte during the Li Deinsertion–Insertion Processes. Chem. Mater. 29, 4768–4774 (2017).
Cao, D. et al. Stable Thiophosphate-Based All-Solid-State Lithium Batteries through Conformally Interfacial Nanocoating. Nano Lett. 20, 1483–1490 (2020).
Garcia‐Mendez, R., Smith, J. G., Neuefeind, J. C., Siegel, D. J. & Sakamoto, J. Correlating Macro and Atomic Structure with Elastic Properties and Ionic Transport of Glassy Li2S‐P2S5 (LPS) Solid Electrolyte for Solid‐State Li Metal Batteries. Adv. Energy Mater. 10, 2000335 (2020).
Kong, S. T. et al. Structural Characterisation of the Li Argyrodites Li7PS6 and Li7PSe6 and their Solid Solutions: Quantification of Site Preferences by MAS-NMR Spectroscopy. Chem. Eur. J. 16, 5138–5147 (2010).
Gobet, M., Greenbaum, S., Sahu, G. & Liang, C. Structural Evolution and Li Dynamics in Nanophase Li3PS4 by Solid-State and Pulsed-Field Gradient NMR. Chem. Mater. 26, 3558–3564 (2014).
Neuberger, S., Culver, S. P., Eckert, H., Zeier, W. G. & Günne, J. S. auf der. Refinement of the Crystal Structure of Li4P2S6 Using NMR Crystallography. Dalton Trans. 47, 11691–11695 (2018).
Wang, Y. et al. X-ray Photoelectron Spectroscopy for Sulfide Glass Electrolytes in the Systems Li2S–P2S5 and Li2S–P2S5–LiBr. J. Ceram. Soc. Japan 124, 597–601 (2016).
Wenzel, S. et al. Interphase formation and degradation of charge transfer kinetics between a lithium metal anode and highly crystalline Li7P3S11 solid electrolyte. Solid State Ionics 286, 24–33 (2016).
Dietrich, C. et al. Spectroscopic Characterization of Lithium Thiophosphates by XPS and XAS – a Model to Help Monitor Interfacial Reactions in All-Solid-State Batteries. Phys. Chem. Chem. Phys. 20, 20088–20095 (2018).
Sang, L. et al. Understanding the Effect of Interlayers at the Thiophosphate Solid Electrolyte/Lithium Interface for All-Solid-State Li Batteries. Chem. Mater. 30, 8747–8756 (2018).
Dewald, G. F. et al. Experimental Assessment of the Practical Oxidative Stability of Lithium Thiophosphate Solid Electrolytes. Chem. Mater. 31, 8328–8337 (2019).
Walther, F. et al. Visualization of the Interfacial Decomposition of Composite Cathodes in Argyrodite-Based All-Solid-State Batteries Using Time-of-Flight Secondary-Ion Mass Spectrometry. Chem. Mater. 31, 3745–3755 (2019).
Li, Y. et al. Stable and Flexible Sulfide Composite Electrolyte for High-Performance Solid-State Lithium Batteries. ACS Appl. Mater. Interfaces 12, 42653–42659 (2020).
Liu, Z. et al. X-ray Photoelectron Spectroscopy Probing of the Interphase between Solid-State Sulfide Electrolytes and a Lithium Anode. J. Phys. Chem. C 124, 300–308 (2020).
Walther, F. et al. Influence of Carbon Additives on the Decomposition Pathways in Cathodes of Lithium Thiophosphate-Based All-Solid-State Batteries. Chem. Mater. 32, 6123–6136 (2020).
Banerjee, A. et al. Revealing Nanoscale Solid–Solid Interfacial Phenomena for Long-Life and High-Energy All-Solid-State Batteries. ACS Appl. Mater. Interfaces 11, 43138–43145 (2019).
Li, M. et al. Electrochemically Primed Functional Redox Mediator Generator from the Decomposition of Solid State Electrolyte. Nat. Commun. 10, 1890 (2019).
Li, X. et al. Unravelling the Chemistry and Microstructure Evolution of a Cathodic Interface in Sulfide-Based All-Solid-State Li-Ion Batteries. ACS Energy Lett. 4, 2480–2488 (2019).
Ye, L. et al. Toward Higher Voltage Solid-State Batteries by Metastability and Kinetic Stability Design. Adv. Energy Mater. 10, 2001569 (2020).
Carbone, M. R., Topsakal, M., Lu, D. & Yoo, S. Machine-Learning X-ray Absorption Spectra to Quantitative Accuracy. Phys. Rev. Lett. 124, 156401 (2020).
Pascal, T. A. et al. X-ray Absorption Spectra of Dissolved Polysulfides in Lithium–Sulfur Batteries from First-Principles. J. Phys. Chem. Lett. 5, 1547–1551 (2014).
Pascal, T. A., Wujcik, K. H., Wang, D. R., Balsara, N. P. & Prendergast, D. Thermodynamic Origins of the Solvent-Dependent Stability of Lithium Polysulfides from First Principles. Phys. Chem. Chem. Phys. 19, 1441–1448 (2017).
Zhang, W. et al. Multi-Stage Structural Transformations in Zero-Strain Lithium Titanate Unveiled by In Situ X-ray Absorption Fingerprints. J. Am. Chem. Soc. 139, 16591–16603 (2017).
Wang, D. R. et al. Rate Constants of Electrochemical Reactions in a Lithium-Sulfur Cell Determined by Operando X-ray Absorption Spectroscopy. J. Electrochem. Soc. 165, A3487 (2018).
Singh, H. et al. Identification of Dopant Site and Its Effect on Electrochemical Activity in Mn-Doped Lithium Titanate. Phys. Rev. Mater. 2, 125403 (2018).
Yan, D. et al. Ultrathin Amorphous Titania on Nanowires: Optimization of Conformal Growth and Elucidation of Atomic-Scale Motifs. Nano Lett. 19, 3457–3463 (2019).
Zhang, W. et al. Kinetic Pathways of Ionic Transport in Fast-Charging Lithium Titanate. Science 367, 1030–1034 (2020).
Qu, X. et al. Resolving the Evolution of Atomic Layer-Deposited Thin-Film Growth by Continuous In Situ X-Ray Absorption Spectroscopy. Chem. Mater. https://doi.org/10.1021/acs.chemmater.0c04547 (2021).
Taillefumier, M., Cabaret, D., Flank, A.-M. & Mauri, F. X-ray Absorption Near-Edge Structure Calculations with the Pseudopotentials: Application to the K edge in Diamond and α-quartz. Phys. Rev. B 66, 195107 (2002).
Prendergast, D. & Galli, G. X-Ray Absorption Spectra of Water from First Principles Calculations. Phys. Rev. Lett. 96, 215502 (2006).
Gougoussis, C., Calandra, M., Seitsonen, A. P. & Mauri, F. First-principles Calculations of X-ray Absorption in a Scheme Based on Ultrasoft Pseudopotentials: from α-quartz to High-Tc Compounds. Phys. Rev. B 80, 075102 (2009).
Giannozzi, P. et al. QUANTUM ESPRESSO: a Modular and Open-source Software Project for Quantum Simulations of Materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Karsai, F., Humer, M., Flage-Larsen, E., Blaha, P. & Kresse, G. Effects of Electron-Phonon Coupling on Absorption Spectrum: K Edge of Hexagonal Boron Nitride. Phys. Rev. B 98, 235205 (2018).
Vinson, J., Rehr, J. J., Kas, J. J. & Shirley, E. L. Bethe-Salpeter Equation Calculations of Core Excitation Spectra. Phys. Rev. B 83, 115106 (2011).
Gulans, A. et al. exciting: a full-potential all-electron package implementing density-functional theory and many-body perturbation theory. J. Phys.: Condens. Matter 26, 363202 (2014).
Bellafont, N. P., Viñes, F., Hieringer, W. & Illas, F. Predicting Core Level Binding Energies Shifts: Suitability of the Projector Augmented Wave Approach as Implemented in Vasp. J. Comput. Chem. 38, 518–522 (2017).
Hamann, D. R. & Muller, D. A. Absolute and Approximate Calculations of Electron-Energy-Loss Spectroscopy Edge Thresholds. Phys. Rev. Lett. 89, 126404 (2002).
Gougoussis, C. et al. Intrinsic Charge Transfer Gap in NiO from Ni K-Edge X-ray Absorption Spectroscopy. Phys. Rev. B 79, 045118 (2009).
England, A. H. et al. On the Hydration and Hydrolysis of Carbon Dioxide. Chem. Phys. Lett. 514, 187–195 (2011).
Guo, H., Wang, Q., Urban, A. & Artrith, N. Artificial Intelligence-Aided Mapping of the Structure–Composition–Conductivity Relationships of Glass–Ceramic Lithium Thiophosphate Electrolytes. Chem. Mater. 34, 6702–6712 (2022).
Ong, S. P. et al. Python Materials Genomics (Pymatgen): A Robust, Open-Source Python Library for Materials Analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Talirz, L. et al. Materials Cloud, a Platform for Open Computational Science. Sci. Data 7, 299 (2020).
Kresse, G. & Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186 (1996).
Ceperley, D. M. & Alder, B. J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 45, 566–569 (1980).
Artrith, N., Urban, A. & Ceder, G. Constructing First-Principles Phase Diagrams of Amorphous LixSi using Machine-Learning-Assisted Sampling with an Evolutionary Algorithm. J. Chem. Phys. 148, 241711 (2018).
Lacivita, V., Artrith, N. & Ceder, G. Structural and Compositional Factors That Control the Li-Ion Conductivity in LiPON Electrolytes. Chem. Mater. 30, 7077–7090 (2018).
Artrith, N. & Urban, A. An Implementation of Artificial Neural-Network Potentials for Atomistic Materials Simulations: Performance for TiO2. Comput. Mater. Sci. 114, 135–150 (2016).
Artrith, N., Urban, A. & Ceder, G. Efficient and Accurate Machine-Learning Interpolation of Atomic Energies in Compositions with Many Species. Phys. Rev. B 96, 014112 (2017).
Miksch, A. M., Morawietz, T., Kästner, J., Urban, A. & Artrith, N. Strategies for the Construction of Machine-Learning Potentials for Accurate and Efficient Atomic-Scale Simulations. Mach. Learn.: Sci. Technol. 2, 031001 (2021).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Rad 12, 537–541 (2005).
Guo, H. et al. Simulated sulfur K-edge X-ray absorption spectroscopy database of lithium thiophosphate solid electrolytes. Materials Cloud https://doi.org/10.24435/materialscloud:6z-qm (2023).
Acknowledgements
We acknowledge financial support by the U.S. Department of Energy (DOE) Office of Energy Efficiency and Renewable Energy, Vehicle Technologies Office, Contract No. DE-SC0012704. The research used the theory and computational resources of the Center for Functional Nanomaterials and Beamlines 7-ID-2 and 8-BM of NSLS-II, which are the U.S. DOE Office of Science User Facilities, and the Scientific Data and Computing Center, a component of the Computational Science Initiative, at Brookhaven National Laboratory under the Contract No. DE-SC0012704. We also acknowledge computing resources from Columbia University’s Shared Research Computing Facility project, which is supported by NIH Research Facility Improvement Grant 1G20RR030893-01, and associated funds from the New York State Empire State Development, Division of Science Technology and Innovation (NYSTAR) Contract C090171, both awarded April 15, 2010. Commercial equipment, instruments, or materials are identified in this paper to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Author information
Authors and Affiliations
Contributions
H.G.: structure selection, DFT calculations, benchmarking and parameter optimization of the EXC simulations, workflow conception, writing – initial draft and editing. M.R.C.: workflow implementation, DFT and spectral calculations, analysis, writing – review and editing. C.C., Y.D., S.B., C.W.: experimental XAS data acquisition. J.Q.: workflow implementation. F.W.: project conception, supervision. N.A.: project conception, structure selection, workflow conception and implementation, writing – review and editing. A.U.: conception, implementation of the workflow, writing – review and editing. D.L.: project conception, supervision, writing – review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, H., Carbone, M.R., Cao, C. et al. Simulated sulfur K-edge X-ray absorption spectroscopy database of lithium thiophosphate solid electrolytes. Sci Data 10, 349 (2023). https://doi.org/10.1038/s41597-023-02262-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02262-4
