
Abstract
The ectoparasitoid wasp Theocolax elegans is a cosmopolitan and generalist pteromalid parasitoid of several major storage insect pests, and can effectively suppress a host population in warehouses. However, little molecular information about this wasp is currently available. In this study, we assembled the genome of T. elegans using PacBio long-read sequencing, Illumina sequencing, and Hi-C methods. The genome assembly is 662.73 Mb in length with contig and scaffold N50 values of 1.15 Mb and 88.8 Mb, respectively. The genome contains 56.4% repeat sequences and 23,212 protein-coding genes were annotated. Phylogenomic analyses revealed that T. elegans diverged from the lineage leading to subfamily Pteromalinae (Nasonia vitripennis and Pteromalus puparum) approximately 110.5 million years ago. We identified 130 significantly expanded gene families, 34 contracted families, 248 fast-evolving genes, and 365 positively selected genes in T. elegans. Additionally, 260 olfactory receptors and 285 venom proteins were identified. This genome assembly provides valuable genetic bases for future investigations on evolution, molecular biology and application of T. elegans.
Similar content being viewed by others

Background & Summary
Postharvest infestation by insect pests largely affects both the quality and quantity of stored grains and related commodities1,2. Infesting insects not only consume grains, but also transport storage fungi and bacteria3. Quantitative and qualitative losses from insect pests during storage are estimated to amount to 20–30%1. To minimize losses, fumigation has been widely used worldwide. However, such massive application of pesticides has resulted in resistance development among pests as well as negative effects on nontarget organisms, human health and the environment4,5. In comparison, biological control using natural enemies presents a safer and more biorational approach. Currently, parasitoid wasps from Pteromalidae, Bethylidae, Braconidae and Ichneumonidae have been reported as natural enemies of many important stored product pests, and several species have been sensibly utilized in the control of insect pests in storehouses6,7,8. Parasitoids suppress host immunity by injecting virulence factors including venom during oviposition, and the progeny consume and eventually kill the hosts, effectively, sustainably and safely controlling the host population9,10.
The ectoparasitoid wasp Theocolax elegans (Westwood) is a cosmopolitan and generalist pteromalid parasitoid of many major storage insect pests11. Augmentative releases of T. elegans can effectively diminish more than 90% of the Rhyzopertha dominica population in stored wheat as well as insect fragments in wheat flour12,13. Compared to most parasitoid wasps whose hosts are restricted to one or more genera from the same order, T. elegans has a broader host spectrum, and is capable of parasitizing hosts of two orders: coleopteran beetles and lepidopteran moth13,14. Previous studies have suggested that variation in the olfactory receptor (OR) and venom repertoire among parasitoid wasps is associated with changing host range15,16,17. The highly dynamic evolution of OR genes, which discriminate odour molecules derived from hosts and host habitats, is crucial for parasitoids to locate different hosts17,18,19. Additionally, venom proteins, which regulate host immunity, development and metabolism, likely evolve quickly and adopt novel functions in response to different host species15,16,20. Although the biology of T. elegans has been studied, little genetic information is presently available, which constrains its desirability and application as a biocontrol agent.
Here, we report a high-quality chromosome-level genome assembly of T. elegans using a combination of PacBio long-read sequencing, Illumina short-read sequencing and Hi-C technology. The final assembly is 662.73 Mb in length with scaffold N50 of 88.8 Mb and 94.3% completeness, providing an excellent genomic resource for subsequent research on T. elegans. The genome contains 56.4% repeat sequences and 23,212 protein-coding genes were annotated. To elucidate the evolutionary position of T. elegans, phylogenomic analyses revealed that this wasp diverged from the lineage leading to subfamily Pteromalinae (Nasonia vitripennis and Pteromalus puparum) approximately 110.5 million years ago. To examine genes that might evolve adaptively, we detected 130 significantly expanded, 34 contracted families, 248 fast-evolving genes, and 365 positively selected genes in T. elegans. We also identified 260 OR and 285 venom proteins. This genome provides valuable resources and insight into the fundamental aspects of evolution, molecular biology and application of T. elegans.
Methods
Sampling and sequencing
T. elegans parasitoid wasps were initially collected from farm-stored wheat in the experimental farmlands of Huajiachi campus, Zhejiang University (Hangzhou, China) and were reared on larvae of the rice weevil host Sitophilus oryza in the laboratory for at least one year before genome sequencing. The genomic DNA of approximately 150 male yellow pupae was isolated using sodium dodecyl sulfate (SDS) and proteinase K digestion, followed by phenol‒chloroform extraction. Two libraries were constructed for genome sequencing. A short-read sequencing library with an insert size of 400 bp was prepared using a Truseq Nano DNA HT Sample Preparation Kit (Illumina, USA) and sequenced using the Illumina Hiseq X Ten platform at GrandOmics Biosciences Co., Ltd. (Wuhan, China). For the 20 kb long-read library, a PacBio SMRTbell library was constructed using SMRTbell Express Template Prep Kit 2.0 (PacBio, USA) and sequenced using Single-Molecule Real-Time (SMRT) cells with the PacBio Sequel sequencer in GrandOmics Biosciences Co., Ltd. (Wuhan, China). A total of 23.7 Gb and 64.33 Gb of clean data were generated from the Illumina paired-end and PacBio libraries, respectively.
For Hi-C sequencing, the library was constructed following the standard protocol as described by Belton et al. with some modifications21. In brief, 150 male yellow pupae were ground into pieces and cross-linked by incubating in 2% formaldehyde solution. Nuclei were isolated and digested with MboI, followed by marking with biotin-14-dCTP. The ligated DNA was sheared into fragments of 300 to 600 bp, and then blunt-end repaired and A-tailed, followed by purification through biotin–streptavidin-mediated pull down. The Hi-C library was amplified by PCR (12–14 cycles) and eventually quantified and sequenced using the Illumina HiSeq 2000 platform at Annoroad Gene Technology Co., Ltd. (Beijing, China).
For transcriptome sequencing, 3–5 d-old larvae (male and mixed-sexed), yellow pupae (male and female), 2–3 d-old adults (male and female) and venom glands from 3–5 d-old female adults of T. elegans were collected separately with three replicates. RNA was extracted using TRIzol® Reagent (Invitrogen, USA) according to the manufacturer’s protocol. RNA-Seq libraries were prepared using TruSeq RNA Sample Prep Kit (Illumina, USA) and sequenced using the Illumina HiSeq X Ten platform at GrandOmics Biosciences Co., Ltd. (Wuhan, China).
Genome assembly
The genome size of T. elegans was first inferred from a 17-mer distribution using Illumina paired-end reads22, and the genome size was estimated to be 752.6 Mb (Supplementary Fig. S1). For de novo genome assembly, PacBio long reads corrected with Falcon (v1.8.7) were assembled to generate an initial assembly by Wtdbg (v1.2.8) (https://github.com/ruanjue/wtdbg-1.2.8)23. The initial assembly was then error-corrected with SMRTlink (v4.0) (https://www.pacb.com/support/software-downloads/) and polished with Illumina reads using Pilon (v1.22)24. This approach generated a 662.63 Mb assembly, with 2,824 contigs and a contig N50 of 1.15 Mb (Table 1).
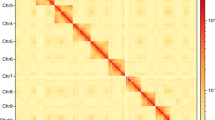
The Hi-C sequencing reads were mapped to the assembled genome using bowtie2 (v2.3.2)25. Uniquely valid paired‐end reads were retained for downstream analysis. Valid interaction pairs were identified using HiC-Pro (v2.7.8)26, and the scaffolds were anchored, ordered and oriented to pseudochromosomes with LACHESIS (https://github.com/shendurelab/LACHESIS)27. A heatmap was drawn to illustrate the interaction of each chromosome. The Hi-C technique oriented and anchored 931 scaffolds (91.9% of the whole genome assembly) to seven chromosomes (Supplementary Fig. S2). The chromosome-level genome assembly was 662.73 Mb in length with 1,900 scaffolds and a scaffold N50 of 88.8 Mb (Table 1). BUSCO (v3.0.1) was used to assess the completeness of genome assembly with the insect protein set (insecta_odb9)28. The results showed that 94.3% of BUSCO genes were successfully detected, of which 93.2% were single-copy and 1.1% duplicates (Table 1).
Genome annotation
A combined de novo and homology-based search was applied to identify repetitive sequences in the T. elegans genome. The de novo repeat library was built with default parameters using RepeatModeler (v2.0.1)29. RepeatMasker (v4.0.7)30 was used to annotate the repetitive elements via searching against the RepBase database (v16.02)31 and the de novo repeat library. We identified 373.7 Mb of repetitive sequences, accounting for 56.4% of the assembled genome, which was the largest ratio among the 10 hymenopteran insects including two pteromalid parasitoid wasps, Nasonia vitripennis (41.66%) and Pteromalus puparum (42.67%) (Fig. 1a). Specifically, four classes of transposable elements (TEs) including long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), DNA elements (DNAs) and short interspersed nuclear elements (SINEs), comprise 15.08%, 7.28%, 5.5%, and 0.53% of the T. elegans genome respectively (Table 2). TE landscapes based on Kimura distance values of four closely-related parasitoid wasps were calculated using RepeatMasker built-in scripts (calcDivergenceFromAlign.pl). The results indicated a recent transposition burst of LTRs, LINEs and DNAs in the three pteromalid parasitoids T. elegans, N. vitripennis and P. puparum (Fig. 1b). Moreover, a recent burst of SINEs was observed in T. elegans.

Transposable elements (TE) characteristics in the ectoparasitoid Theocolax elegans genome. (a) Comparison of TE content among 10 hymenopteran genomes. Tel, T. elegans; Nvi, Nasonia vitripennis; Ppu, Pteromalus puparum; Tpr, Trichogramma pretiosum; Far, Fopius arisanus; Mde, Microplitis demolitor; Ame, Apis mellifera; Ace, Atta cephalotes; Oab, Orussus abietinus; Aro, Athalia rosae. (b) Interspersed landscape of four major transposable elements (LINEs, DNAs, SINEs and LTRs). The y-axis shows the genome percent, and the x-axis shows the level of Kimura substitution (CpG adjusted) of each repeat family.
After masking repeat sequences, protein homologue searching, transcriptome sequencing and de novo prediction were integrated to predict protein-coding genes using the Optimized Maker-Based Insect Genome Annotation (OMIGA) pipeline32. For homology searching, all invertebrate protein sequences downloaded from the National Center for Biotechnology Information (NCBI) RefSeq database were aligned to the T. elegans genome using exonerate (v2.2.0)33. For transcriptome-based prediction, clean reads filtered by Trimmomatic (v0.36)34 were mapped to the genome assembly using HISAT2 (v2.1)35 and assembled into transcripts using StringTie (v1.3.4c)36. For de novo prediction, three prediction programs including Augustus (v3.1)37, SNAP (v2006-07-28)38 and GeneMark-ET (v4.21)39 were used. All gene evidences identified from the above three approaches were combined by MAKER (v2.31)40 into a weighted and nonredundant consensus of gene structures with default parameters. The predicted protein-coding genes were functionally annotated by searching against the Swiss-Prot and NCBI Nr databases using blastp (v2.8.1). Conserved domains of proteins were analysed against the Pfam (v32.0) database with HMMER (v3.3.2)41. The genes were mapped to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using BlastKOALA (https://www.kegg.jp/blastkoala/)42, and Gene Ontology (GO) annotation was performed using Blast2GO (v5.2)43. Ultimately, we predicted 23,212 protein-coding genes in the genome of T. elegans, 20,986 (90.4%) of which were successfully annotated in at least one database (Table 3).
Orthologue and phylogenetic analyses
Protein sequences of 10 hymenopteran species were used for comparative genomics and phylogenomic analyses: T. elegans (this study), N. vitripennis (OGS244), P. puparum45, Trichogramma pretiosum (RefSeq assembly accession: GCF_000599845.2), Fopius arisanus (GCF_000806365.1), Microplitis demolitor (GCF_000572035.2), Apis mellifera (GCF_003254395.2), Atta cephalotes (GCF_000143395.1), Orussus abietinus (GCF_000612105.2) and Athalia rosae (GCF_000344095.2). The longest transcript of each gene was retained for orthologue and phylogenetic analyses. OrthoFinder (v2.5.1)46 with default settings was used to identify orthologous and paralogous genes. A total of 3,199 single-copy orthogroups were identified and extracted for phylogenetic analysis (Fig. 2). All protein sequences were aligned with MAFFT (v7.123b)47 and trimmed by trimAl (v1.4.rev22)48. The sequences were concatenated into a supergene sequence and used for phylogenetic analysis. A phylogenetic tree was constructed by maximum likelihood (ML) method using IQ-TREE (v2.1.2)49, with 1000 ultrafast bootstrap replicates. The best model (JTT + F + R6) was determined by ModelFinder50 and the basal hymenopteran herbivore A. rosae was used as an outgroup51. Phylogenetic inference indicated the phylogenetic location of T. elegans in Pteromalidae along with two Pteromalinae species (N. vitripennis and P. puparum). The four chalcidoids (T. elegans, N. vitripennis, P. puparum and T. pretiosum) cluster together, with two braconids (F. arisanus and M. demolitor) as a sister group. The phylogenetic arrangement was consistent with previous studies using inference based on transcriptomic data51,52. Divergence times were estimated using the MCMCTree program in the PAML package (v4.9)53 based on protein sequences. Three calibration time points were used for calibration purposes: Chalcidoidea, 105–159 million years ago (mya), Apocrita, 203–276 mya and Orussoidea + Apocrita, 211–289 mya45,51. The results suggest that T. elegans diverged from the lineage leading to subfamily Pteromalinae (N. vitripennis and P. puparum) approximately 110.5 mya (Fig. 2). Cafe (v5)54 software was used to analyse gene family evolution in T. elegans with default parameters, and gene families inferred from OrthoFinder and estimated divergence times were used as inputs. The results suggested 511 expanded and 1,843 contracted gene families in T. elegans (Fig. 2). Among them, 130 and 34 gene families experienced significant expansion and contraction events (P < 1E-5), respectively (see table deposited at Figshare55).

Phylogenetic and comparative genomic analyses of the ectoparasitoid Theocolax elegans. To the left is the maximum likelihood phylogenetic tree built from 3,199 concatenated single-copy orthologous groups from T. elegans and other nine hymenopterans using IQ-TREE. The basal hymenopteran Athalia rosae was used as an outgroup. All nodes received 100% bootstrap support. Numbers of expanded (red) and contracted (green) gene families are shown on the branches. To the right is the total gene counts of different types of orthologous groups in each genome. “1:1:1” indicates universal single-copy genes present in all species; “N:N:N” indicates other universal genes; “Chalcidoidea specific” indicates common unique genes in the four Chalcidoidea species; “Species specific” represents species specific genes with more than one copies in the genome; “Unassigned genes” indicates species-specific genes with only one copy in the genome; “Others” indicates remaining genes.
Gene evolution analyses
To detect genes that might be related to adaptive evolution in T. elegans, fast-evolving genes (FEGs) and positively selected genes (PSGs) were inferred by ratio of non-synonymous to synonymous substitutions (dN/dS, ω) analysis of each single-copy gene, using the branch model and branch-site model by codeml in PAML (v4.9e)53, respectively. Multiple protein sequence alignments were converted to the corresponding coding sequence (CDS) alignments using ParaAT (v2.0)56. FEGs were identified by comparing the null model (model = 0) with the alternative model (model = 2). The likelihood ratio test (LRT) was used to discriminate significance between the two models, and significance was further adjusted by the FDR method. Genes with higher ω values in the branch of T. elegans than in the background branches and FDR-adjusted P values less than 0.05 were considered FEGs of T. elegans. Additionally, PSGs at the single-codon level were identified by comparing null Model A (NSsites = 2, model = 2, fix_omega = 1) with Model A (NSsites = 2, model = 2, fix_omega = 0). Probabilities of amino acid positions with ω > 1 were estimated by the Bayes empirical Bayes (BEB) test implemented in PAML. Genes with positive selection sites and FDR-adjusted P values less than 0.05 were identified as PSGs. In total, we identified 248 FEGs (see table deposited at Figshare57) and 365 PSGs (see table deposited at Figshare58) in T. elegans, with 57 genes overlapping.
Annotation and phylogenetic analysis of olfactory receptor genes
To compare the olfactory receptor (OR) repertoire among parasitoid wasps, we annotated OR genes of T. elegans and eight other parasitoid wasps with high-quality genome assemblies, including N. vitripennis, P. puparum, Copidosoma floridanum (GCF_000648655.2), T. pretiosum, Telenomus remus (GCA_020615435.1), Cotesia chilonis (GCA_018835575.1), Diadromus collaris (GCA_009394715.1) and Gonatopus flavifemur (GCA_018340375.1), with the InsectOR pipeline (https://github.com/sdk15/insectOR)59. Well-annotated OR protein sequences from N. vitripennis, A. mellifera, M. mediator, Megachile rotundata, and Bombus impatiens were used as queries to search against the nine parasitoid wasp genomes. Predicted OR proteins with lengths greater than or equal to 300 amino acids and with the 7tm_6 domain predicted by InterProScan (v5.48-83.0)60 were defined as intact OR genes and used for further analyses61. All predicted protein sequences were aligned using MAFFT (v7.123b)47 and then trimmed by trimAl (v1.4.rev22)48. Phylogenetic analysis of OR proteins was performed using ML methods with the JTT + F + G4 model determined by ModelFinder50 in IQ-TREE (v2.1.2)43. Statistical support for the phylogenetic tree was assessed by ultrafast bootstrap analysis using 1000 replicates. The clade for odourant receptor coreceptor (Orco) proteins was applied as the outgroup for the phylogenetic tree. Overall, 1,268 intact OR genes were identified in nine parasitoid wasps, and extensive variation in the size of OR repertoires was observed, ranging from 46 in Telenomus remus to 260 in T. elegans (Fig. 3a,b). Gene gain and loss events were predicted using NOTUNG (v2.9.1.5)62 and mapped onto a species cladogram of nine parasitoid wasps inferred by former studies51,52. The results indicated that 133 and 120 OR gene gain and loss events occurred in T. elegans (Fig. 3b). OR genes were then further classified into 19 different subfamilies based on statistical support (bootstrap values higher than 70%) and subfamily definition of N. vitripennis OR genes in previous studies61,63. Among the nine parasitoid wasps, T. elegans has the most abundant OR genes of six subfamilies including Z (45 OR genes), E (35), F (34), T (25), V (22) and U (6), which comprise 64.2% of the OR repertoire (Fig. 3b).

Phylogenetic analysis of olfactory receptor (OR) proteins of nine parasitoid wasps. (a) Maximum likelihood OR protein tree with branches coloured by nine parasitoid wasp species. Scale bar represents 0.5 mean substitutions per site. (b) To the left is a cladogram of nine parasitoid wasps showing estimated number of OR gene gain and loss events along branches and estimated size of ancestral and extant species OR repertoires highlighted in grey. To the right is a bar chart showing the number of each OR subfamilies.
Identification of venom proteins
Venom proteins of T. elegans were identified using a proteo-transcriptomic approach. Approximately 100 venom glands from 3–5 d-old female parasitoids were collected separately with three replicates. RNA-Seq libraries were prepared and sequenced as mentioned above. Gene expression levels represented by transcripts per kilobase million (TPM) were estimated using RSEM (v1.3.3)64. Proteomic analysis was performed as described previously, with some modifications65,66. In brief, approximately 200 venom reservoirs were centrifuged at 12,000 × g for 10 min, and the supernatant was collected and digested with trypsin. The peptides were loaded onto a Thermo Scientific EASY Column (2 cm*100 μm, 5 μm-C18) and then separated on a Thermo Scientific EASY column (75 μm*100 mm, 3 μm-C18). Buffer A was water with 0.1% formic acid; buffer B was 84% acetonitrile with 0.1% formic acid. The buffer B gradient was as follows: 0–110 min, from 0% to 55%; 110–115 min, from 55% to 100%; 115–120 min, 100%. Resulting MS/MS spectra were searched against protein sequences using MaxQuant (v2.0.3.1)67 with the filtration criterion of FDR ≤ 0.01. To minimize false-positive, genes with reliable transcript levels (TPM ≥ 50) in the venom gland transcriptome and detected in proteomics with more than two unique peptides were defined as venom genes of T. elegans68. In total, 285 venom proteins were identified (see table deposited at Figshare69). These proteins were further categorized into enzymes (122), protease inhibitors (3), recognition and binding proteins (57), others (62) and unknown (41). The most abundant category was “enzymes” (42.8%), including serine proteases, lipase, and metalloproteases; the second most abundant category was “others” (21.8%), including heat shock proteins, major royal jelly proteins and yellow proteins.
Data Records
Illumina, PacBio and Hi-C data for T. elegans genome sequencing are available as BioProject PRJNA868490 (SRA accessions SRR2101098570, SRR2101098471 and SRR2101098272, respectively). Illumina transcriptome data for male larvae, mixed-sexed larvae, male pupae, female pupae, male adults and female adults with three replicates are also available as BioProject PRJNA868490 (SRA accessions SRR21010979 - SRR2101098173,74,75, SRR2101099476, SRR2101099577, SRR2101098378, SRR21010976 - SRR2101097879,80,81, SRR2101097582, SRR2101099383, SRR2101099284, SRR21010989 - SRR2101099185,86,87, SRR21010986 - SRR2101098888,89,90, respectively). Illumina transcriptome data for venom glands with three replicates are available as BioProject PRJNA868589 (SRA accessions SRR21011763 - SRR2101176591,92,93), and mass spectrometry proteomics data are accessible via the PRIDE94 database under accession number of PXD03777495. The genome assembly96, gene CDS97 and protein98 data were deposited in the Figshare database. In addition, the genome assembly has been submitted to NCBI under accession number GCA_026168455.199.
Technical Validation
DNA quality and concentration were measured using pulse field gel electrophoresis (0.7%) and Qubit 3.0 (Thermo Fisher Scientific, USA), respectively. The integrity and quantity of RNA were evaluated using an Agilent 2100 Bioanalyzer (Agilent, USA). High-quality DNA and RNA were used for library preparation and sequencing.
Code availability
Software parameters of genome assembly: default parameters for HiC-Pro and BUSCO. Falcon: length_cutoff = 13000 length_cutoff_pr = 14000; pa_HPCdaligner_option = -v -B188 -M24 -t12 -e.75 –k18 -w8 –h280 –l2800 -s1000, ovlp_HPCdaligner_option = -v –B128 –h180 -e.96 –k17 –l2800 -s1000.
Wtdbg:–tidy-reads 5000 -fo dbg -k 0 -p 19 -S 3 -E 5–rescue-low-cov-edges–aln-noskip; wtdbg-cns -c 3; kbm-1.2.8 -k 0 -p 19 -S 4 -O 0; map2dbgcns; wtdbg-cns -k 13 -c.
SMRTlink:–bam–bestn 5–minMatch 18–nproc 6–minSubreadLength 1000–minAlnLength 500–minPctSimilarity 70–minPctAccuracy 70–hitPolicy randombest–randomSeed 1.
LACHESIS: CLUSTER MIN RE SITES = 100; CLUSTER MAX LINK DENSITY = 2.5; CLUSTER NONINFORMATIVE RATIO = 1.4; ORDER MIN N RES IN TRUNK = 60; ORDER MIN N RES IN SHREDS = 60.
Software parameters of genome annotation: default parameters for SNAP, GeneMark-ET, MAKER, Trimmomatic, HISAT2 and StringTie.
exonerate:–model protein2genome –percent 50–score 100–minintron 20–maxintron 20000.
Augustus:–species = nasonia–noInFrameStop = true–gff3 = on–strand = both.
RepeatModeler: -engine ncbi -LTRStruct.
RepeatMasker -a.
blasp: -e 1e-5.
HMMER: -E 1e-5.
BlastKOALA: Eukaryotes for taxonomy group and family_eukaryotes for KEGG GENES database file to be searched.
Software parameters of orthologue and phylogenetic analyses: default parameters for OrthoFinder, MCMCTree.
MAFFT:–auto.
trimAl: -automated1.
IQ-TREE: -m JTT + F + R6 -B 1000 -T AUTO.
Cafe: -p.
Software parameters of gene evolution analyses: default parameters for codeml.
ParaAT: -f paml -m mafft.
Software parameters of annotation and phylogenetic analysis of olfactory receptor genes: default parameters for the InsectOR pipeline.
MAFFT:–maxiterate 1000–localpair.
trimAl: -automated1.
IQ-TREE: -m JTT + F + G4 -B 1000 -T AUTO.
Software parameters of venom protein identification: default parameters for MaxQuant.
RSEM:–bowtie2.
Custom scripts were provided at personal GitHub (https://github.com/xiaoshan40/scripts), including scripts to retrieve the longest protein and CDS sequences for each gene (get_longest_protein_and_cds.pl), to concatenate aligned protein sequences to a supergene sequence (concatenate_aligned_sequences.pl), to automatically perform gene evolution analyses (run_codeml.pl) and to extract results from the original branch and branchsite model output files by codeml (get_paml_branch_result.pl and get_paml_branchsite_result.pl).
References
Taddese, M. et al. Assessment of quantitative and qualitative losses of stored grains due to insect infestation in Ethiopia. J Stored Prod Res 89, 101689 (2020).
Meneghetti, V. L. et al. Evaluation of losses and quality maintenance of wheat during storage in a commercial unit in Brazil. J Sci Food Agric 102, 1569–1575 (2022).
Setamou, M., Cardwell, K. F., Schulthess, F. & Hell, K. Effect of insect damage to maize ears, with special reference to Mussidia nigrivenella (Lepidoptera: Pyralidae), on Aspergillus flavus (Deuteromycetes: Monoliales) infection and aflatoxin production in maize before harvest in the Republic of Benin. J Econ Entomol 91, 433–438 (1998).
Nayak, M. K., Daglish, G. J., Phillips, T. W. & Ebert, P. R. Resistance to the fumigant phosphine and its management in insect pests of stored products: a global perspective. Annu Rev Entomol 65, 333–350 (2020).
Boyer, S., Zhang, H. & Lempérière, G. A review of control methods and resistance mechanisms in stored-product insects. Bull Entomol Res 102, 213–229 (2012).
Phillips, T. W. & Throne, J. E. Biorational approaches to managing stored-product insects. Annu Rev Entomol 55, 375–397 (2010).
Amante, M., Schoeller, M., Suma, P. & Russo, A. Bethylids attacking stored-product pests: an overview. Entomol Exp Appl 163, 251–264 (2017).
Harush, A. et al. Integrated pest management for stored grain: potential natural biological control by a parasitoid wasp community. Insects 12, 1038 (2021).
Moreau, S. J. M. & Asgari, S. Venom proteins from parasitoid wasps and their biological functions. Toxins 7, 2385–2412 (2015).
Haverkamp, A. & Smid, H. M. A neuronal arms race: the role of learning in parasitoid-host interactions. Curr Opin Insect Sci 42, 47–54 (2020).
Adarkwah, C., Obeng-Ofori, D., Opuni-Frimpong, E., Ulrichs, C. & Schöller, M. Predator-parasitoid-host interaction: biological control of Rhyzopertha dominica and Sitophilus oryzae by a combination of Xylocoris flavipes and Theocolax elegans in stored cereals. Entomologia Experimentalis Et Applicata 167, 118–128 (2019).
Flinn, P. W. & Hagstrum, D. W. Augmentative releases of parasitoid wasps in stored wheat reduces insect fragments in flour. J Stored Prod Res 37, 179–186 (2001).
Flinn, P. W. Temperature effects on efficacy of Choetospila elegans (Hymenoptera: Pteromalidae) to suppress Rhyzopertha dominica (Coleoptera: Bostrichidae) in stored wheat. J Econ Entomol 91, 320–323 (1998).
Perez-Benavides, A. L., Hernandez-Baz, F., Gonzalez, J. M. & Riveron, A. Z. Updated taxonomic checklist of Chalcidoidea (Hymenoptera) associated with Bruchinae (Coleoptera: Chrysomelidae). Zootaxa 4638, 301–343 (2019).
Martinson, E. O., Mrinalini, Kelkar, Y. D., Chang, C. H. & Werren, J. H. The evolution of venom by co-option of single-copy genes. Curr Biol 27, 2007–2013 (2017).
Cavigliasso, F., Mathé-Hubert, H., Gatti, J.-L., Colinet, D. & Poirié, M. Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species. Peer Community Journal 1, e51 (2021).
Gauthier, J. et al. Chromosomal scale assembly of parasitic wasp genome reveals symbiotic virus colonization. Commun Biol 4, 104 (2021).
Tang, Q. Olfactory responses of Theocolax elegans (Hymenoptera, Pteromalidae) females to volatile signals derived from host habitats. J Hymenopt 49, 95–109 (2016).
Jongepier, E. et al. Convergent loss of chemoreceptors across independent origins of slave-making in ants. Mol Biol Evol 39, msab305 (2022).
Ye, X. et al. Genomic signatures associated with maintenance of genome stability and venom turnover in two parasitoid wasps. Nat Commun 13, 6417 (2022).
Belton, J.-M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13, 1050–1054 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125 (2013).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 4, 10 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Liu, J., Xiao, H., Huang, S. & Li, F. OMIGA: Optimized Maker-Based Insect Genome Annotation. Mol Genet Genomics 289, 567–573 (2014).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform 6, 31 (2005).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439 (2006).
Korf, I. Gene finding in novel genomes. BMC Bioinform 5, 59 (2004).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42, e119 (2014).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18, 188–196 (2008).
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform 11, 431 (2010).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428, 726–731 (2016).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Rago, A. et al. OGS2: genome re-annotation of the jewel wasp Nasonia vitripennis. BMC Genom 17, 678 (2016).
Ye, X. et al. A chromosome-level genome assembly of the parasitoid wasp Pteromalus puparum. Mol Ecol Resour 20, 1384–1402 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32, 268–274 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14, 587–589 (2017).
Peters, R. S. et al. Evolutionary history of the hymenoptera. Curr Biol 27, 1013–1018 (2017).
Peters, R. S. et al. Transcriptome sequence-based phylogeny of chalcidoid wasps (Hymenoptera: Chalcidoidea) reveals a history of rapid radiations, convergence, and evolutionary success. Mol Phylogenet Evol 120, 286–296 (2018).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2020).
Xiao, S. Rapidly evolving gene families in Theocolax elegans. figshare https://doi.org/10.6084/m9.figshare.21408012 (2023).
Zhang, Z. et al. ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem Biophys Res Commun 419, 779–781 (2012).
Xiao, S. Fast-evolving genes identified in Theocolax elegans. figshare https://doi.org/10.6084/m9.figshare.21408051 (2023).
Xiao, S. Positively selected genes identified in Theocolax elegans. figshare https://doi.org/10.6084/m9.figshare.21408090 (2023).
Karpe, S. D., Tiwari, V. & Ramanathan, S. InsectOR-Webserver for sensitive identification of insect olfactory receptor genes from non-model genomes. PLoS One 16, e0245324 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Legan, A. W., Jernigan, C. M., Miller, S. E., Fuchs, M. F. & Sheehan, M. J. Expansion and accelerated evolution of 9-exon odorant receptors in Polistes paper wasps. Mol Biol Evol 38, 3832–3846 (2021).
Chen, K., Durand, D. & Farach-Colton, M. NOTUNG: a program for dating gene duplications and optimizing gene family trees. J Comput Biol 7, 429–447 (2000).
Robertson, H. M., Gadau, J. & Wanner, K. W. The insect chemoreceptor superfamily of the parasitoid jewel wasp Nasonia vitripennis. Insect Mol Biol 19 (Suppl 1), 121–136 (2010).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12, 323 (2011).
Yang, L. et al. Identification and comparative analysis of venom proteins in a pupal ectoparasitoid, Pachycrepoideus vindemmiae. Front Physiol 11, 9 (2020).
Yan, Z. et al. Insights into the venom composition and evolution of an endoparasitoid wasp by combining proteomic and transcriptomic analyses. Sci Rep 6, 19604 (2016).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11, 2301–2319 (2016).
Zhao, Y. & Lin, Y. H. Whole-cell protein identification using the concept of unique peptides. Genom Proteom Bioinf 8, 33–41 (2010).
Xiao, S. Venom proteins identified in Theocolax elegans. figshare https://doi.org/10.6084/m9.figshare.21408099 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010985 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010984 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010982 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010979 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010980 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010981 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010994 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010995 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010983 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010976 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010977 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010978 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010975 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010993 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010992 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010989 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010990 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010991 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010986 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010987 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21010988 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21011763 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21011764 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21011765 (2022).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Research 50, D543–D552 (2022).
Xiao, S. Venom proteome of the parasitoid wasp Theocolax elegans. PRIDE Archive https://identifiers.org/pride.project:PXD037774 (2022).
Xiao, S. Genome assembly of the parasitoid wasp Theocolax elegans. figshare https://doi.org/10.6084/m9.figshare.21407709 (2022).
Xiao, S. Tele_OGS1.0_mRNA.fasta. figshare https://doi.org/10.6084/m9.figshare.21360066 (2022).
Xiao, S. Tele_OGS1.0_protein.fasta. figshare https://doi.org/10.6084/m9.figshare.21360063 (2022).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_026168455.1 (2022).
Acknowledgements
This work was supported by the Key Program of the National Natural Science Foundation of China (NSFC) (no. 31830074 to G.Y.), the Joint Funds of the National Natural Science Foundation of China (NSFC) (no. U21A20225 to G.Y.) and the Program for Chinese Innovation Team in Key Areas of Science and Technology of Ministry of Science and Technology of the People’s Republic of China (no. 2016RA4008 to G.Y.).
Author information
Authors and Affiliations
Contributions
G.Y. conceived of this project. S.X., X.Y. and Y.Y.participated in the data analysis. S.X., S.W. and F.W. collected the samples. S.X. wrote the manuscript. G.Y., X.Y., Q.F. and S.X. revised the manuscript. All authors have read, revised, and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, S., Ye, X., Wang, S. et al. Genome assembly of the ectoparasitoid wasp Theocolax elegans. Sci Data 10, 159 (2023). https://doi.org/10.1038/s41597-023-02067-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02067-5
