
Abstract
Rose is one of the most important ornamental plants, accounting for one-third of the world’s fresh cut flower market. The vase life refers to the period of a cut flower retaining its appearance in a vase. During this period, the rose was subjected to a variety of abiotic and biotic stresses, resulting in a reduction in the life of cut flowers. Numerous studies have been carried out on cut rose, which proves the effects of various plant hormones on post-harvest dehydration, petal senescence and abscission, disease and vase life of cut rose flowers. In addition, the natural or synthetic hormones or its inhibitor have been successfully used in cut flower preservatives to extend the vase life of rose. However, there is still a lack of systematic and in-depth research on the expression of rose genes related to plant hormone response. Here we analyzed the gene expression changes of the rose flower under treatment of 11 different plant hormones or its inhibitors in order to provide reference for rose studies.
Measurement(s) | Transcripts expression profile |
Technology Type(s) | Illumina HiSeq |
Factor Type(s) | phytohormones |
Sample Characteristic - Organism | Rosa hybrida |
Similar content being viewed by others

Background & Summary
Rose (Rosa sp.) is the most popular flower crop in the world. With its long history of cultivation, the rose has been endowed with cultural connotations in both the eastern and western world. In 2016, the worldwide turnover of cut roses was 4.96 billion euros, accounting for 29.7% of total cut flowers, which was the largest in flower crops (AIPH, 2016).
The vase life refers to the duration of a cut flower retaining its appearance in a vase. It is therefore the most important trait to the ornamental crops that are used as a cut flower, including rose. During the early vase life period, flowers open and appeal flavor, later they start the senescence, dehydration and abscission of petals. In addition, cut flowers are also subject to postharvest diseases, such as gray mold disease caused by the necrotrophic fungus Botrytis cinerea. All these physiological changes occur at a specific stage and in a highly synchronized manner, involving the balance of phytohormones in the flower and up- or down-regulation of numerous genes in various hormones pathways.
Therefore, the effects of plant hormones that extend vase life are major component of floriculture research and have been studied at the physiological and biochemical levels for several decades. Many of chemicals involved in plant hormones and their inhibitors have been added into the preservatives to improve the post-harvesting fresh keeping and vase life of cut rose. In rose, a number of studies have reported that flower opening, petal senescence, dehydration and abscission can be affected by abscisic acid (ABA), cytokinins (CKs), ethylene (ET) and gibberellins (GAs)1,2,3,4,5,6,7,8. In addition, recently, role of brassinosteroids (BRs), jasmonic acid (JA) and ET in rose petal defense against B. cinerea infection have been reported9,10.
However, the global gene expression pattern of rose flowers behind hormonal treatment has not been well-studied yet. To date, the only involved transcriptome data is screening of ethylene responsive genes from rose flowers (SRA045958), which is currently presented in the NCBI Sequence Read Archive (SRA)11.
The information on the molecular mechanism of phytohormones regulating flower traits remains scarce due to the lack of transcriptome. Therefore, transcriptome data from rose petals after different hormone treatments will be useful for studying the expression patterns of hormone-related genes and excavating key genes that regulate flower traits. Using RNA-seq, we recently investigated the transcriptomic dynamics of rose flower under the treatment of eleven natural or synthetic hormones, including auxin, BRs, CKs, GAs, ABA, JA, salicylic acid (SA), ethylene (ET) as well as ethylene inhibitors. We obtained approximately 240 Gb data and dissected the transcriptional network with the aim of exploring the transcriptional variation of rose responses towards those plant hormones. Our data will be useful to all those working with the analysis of rose gene expression.
Methods
Plant materials
Rosa hybrida ‘Samantha’, a classic hybrid tea rose cultivar and frequently used for cut flowers, has a red colour and mild fragrance. The ‘Samantha’ plant was grown under a plastic cover in Changping District (40°139 N, 116°129E), Beijing, China. In Spring of 2017, cut flower samples were harvested at developmental stage 2 of flower opening1.
Exogenous phytohormone treatment
Flowering rose stems were cut into lengths of 30 cm and placed in aqueous solutions of 1-naphthaleneacetic acid (NAA), 2,4-dichlorophenoxyacetic acid (2,4-D), 2,4-epibrassinolide (BR), 6-benzylaminopurine (6-BA), ABA, gibberellic acid 3 (GA3), JA, SA, as well as ethylene inhibitor AgNO3, respectively, for 24 h under the controlled conditions of 22 °C with 30% to 40% relative humidity and 16 h/8 h day/night periods. ‘Samantha’ treated with deionized water were used as the control. For ethylene (ET) and 1-methylcyclopropene (1-MCP) treatment, rose flowers were exposed to ethylene, 1-MCP, or regular air as the control, for 24 h and 1 M NaOH was used to absorb CO2 released by respiration. The detailed information and final concentration of chemical agents were listed in Table 1. For each treatment, three replicates were harvested and 4 flowers were randomly collected for each replicate.
RNA extraction, library construction, and Illumina sequencing
Total RNA was extracted from the outer layer of rose petals using the hot borate reagent following the previous description3. The quality of the RNA was verified by agarose gel electrophoresis, NanoDrop 2000 spectrophotometers (Thermo Fisher Scientific) and Agilent Technologies 2100 Bio-analyzer, all samples QC results were shown in Table 2. The libraries were sequenced on the Illumina HiSeq™ 2500 system (Illumina Inc., San Diego, CA), according to the manufacturer’s instructions. Illumina sequencing was conducted at Novogene, Beijing, China.
Data Records
Our 39 raw data of RNA-seq were deposited into the NCBI database at Sequence Read Archive (SRA) with the accession number PRJNA52266412.
Average gene expression (fragments per kb per million reads, FPKM) information of each experiment was deposited in Gene Expression Omnibus (GEO) in NCBI, number GSE14069613.
Technical Validation
Our 39 raw data were achieved on Illumina HiSeq. The raw data was cleaned by removing the adaptor sequences, reads containing N > 10% (N represents the base cannot be determined), as well as low quality (Qscore < = 5) base which is over 50% of the total base (Table 3)14. FastQC was used to test the quality of 78 paired-end clean data15. We have shown the quality control results of BR-3 clean data as an illustration (Fig. 1). The FPKM (fragments per kb per million reads) of 39 samples representing different treatments were subjected to principal component analysis (PCA), and the clear separation between the treatment and mock was detected (Fig. 2).

Quality control result of BR-3 clean data. (A) Quality score of per position in read. (B) Quality score of mean sequence. (C) GC content distribution. (D) Sequence length distribution.

The principal components analysis (PCA) of RNA-seq data following hormone treatments. (A–I). represented the PCA of 2,4-D, NAA, ABA, BR, 6-BA, GA3, JA, SA and AgNO3 compared with mock treatment (H2O). (J) and (K) represented the PCA of ET and 1-MCP compared with air (ET-CK).
Clean reads were mapped to reference genome Rosa chinensis ‘Old blash’ (RchiOBHm-V2, GCF_002994745.1)16 by default parameters of Tophat2(version 2.1.1). Then use Cufflinks (version 2.2.0) to generate transcriptome with the Tophat2 (version 2.1.1) resulting alignment files, the assemblies were merged with the Cuffmerge, which is included in the Cufflinks package. These merged results provide a uniform basis for calculating gene and transcript expression. Then the merged assembles were provided to Cuffdiff, which calculated expression levels and tested the statistical significance of observed changes.
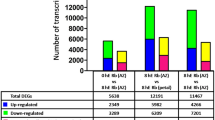
Average gene expression information of each experiment was deposited in Gene Expression Omnibus (GEO) in NCBI, number GSE140696. The differentially expressed genes (DEGs) were analyzed by DESeq (Anders et al., 2013) and defined as genes with |log2 fold change (FC)| ≥ 0.5, and an adjusted P-value < 0.05. The number of DEGs for 11 treatments compared with the control, was shown in Fig. 3.

The number of different expression genes (DEGs) in rose petals under exogenous phytohormone treatments. The DEGs were determined with |log2 fold change (FC)| ≥ 0.5, and an adjusted P-value < 0.05.
In addition, we have analyzed the DEGs under auxin-related plant growth regulators (2,4-D and NAA) and ethylene-related chemicals (ET, 1-MCP and AgNO3). The results showed that 132 overlapped DEGs were identified under 2,4-D and NAA treatments (Fig. 4A). Among the 132 DEGs, 84.09% showed a similar expression pattern under the two different auxin-related plant growth regulators (Fig. 4C). In ET-, 1-MCP- and AgNO3 treatments, there were 245 overlapped DEGs were screened out (Fig. 4B). Although both 1-MCP and AgNO3 are the inhibitors of ET, they work in different ways. We have identified that 81.3% and 26.1% of the 245 DEGs played an opposite expression pattern in 1-MCP and AgNO3 treatment compared with it in ET treatment, respectively (Fig. 4D).

The DEGs among auxin related hormone (2,4-D and NAA), ethylene and its inhibitors (AgNO3 and 1-MCP). (A). Venn diagram depicted the number and overlap DEGs from 2,4-D- and NAA- treatments. (B). Venn diagram depicted the number and overlap DEGs from ET-, AgNO3- and 1-MCP- treatments. (C). The heatmap of overlap DEGs by hierarchical cluster analysis among 2,4-D- and NAA- treatments. (D). The heatmap of overlap DEGs by hierarchical cluster analysis among ET-, AgNO3- and 1-MCP- treatments.
Code availability
The following software and versions were used for quality control and data processing:
1. FastQC: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (version 0.11.9).
2. TopHat2: http://ccb.jhu.edu/software/tophat/index.shtml (version 2.1.1).
3. Cufflinks: http://cole-trapnell-lab.github.io/cufflinks(version 2.2.0).
References
Ma, N., Cai, L., Lu, W., Tan, H. & Gao, J. Exogenous ethylene influences flower opening of cut roses (Rosa hybrida) by regulating the genes encoding ethylene biosynthesis enzymes. Sci China C Life Sci. 48, 434 (2005).
Tan, H. et al. Ethylene-influenced flower opening and expression of genes encoding ETRs, CTRs, and EIN3s in two cut rose cultivars. Postharvest Biol Technol. 40, 97–105 (2006).
Wu, L. et al. An Ethylene-induced regulatory module delays flower senescence by regulating cytokinin content. Plant Physiol. 173, 853–862 (2017).
Xue, J. et al. Expression of ethylene biosynthetic and receptor genes in rose floral tissues during ethylene-enhanced flower opening. J Exp Bot. 59, 2161–2169 (2008).
Ma, N. et al. Rh-PIP2; 1, a rose aquaporin gene, is involved in ethylene-regulated petal expansion. Plant Physiol. 148, 894–907 (2008).
Ma, N. et al. Petal senescence: a hormone view. J Exp Bot. 69, 719–732 (2018).
Lü, P. et al. RhHB1 mediates the antagonism of gibberellins to ABA and ethylene during rose (Rosa hybrida) petal senescence. Plant J. 78, 578–590 (2014).
Luo, J. et al. A DELLA gene, RhGAI1, is a direct target of EIN3 and mediates ethylene-regulated rose petal cell expansion via repressing the expression of RhCesA2. J Exp Bot. 64, 5075–5084 (2013).
Liu, X. et al. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 19, 62 (2018).
Cao, X. et al. A detached petal disc assay and virus-induced gene silencing facilitate the study of Botrytis cinerea resistance in rose flowers. Hortic Res 6, 136 (2019).
Pei, H. et al. An NAC transcription factor controls ethylene-regulated cell expansion in flower petals. Plant Physiol. 163, 775–791 (2013).
NCBI database at Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP186551 (2019).
Liu, X. & Wu, J. Gene Expression Omnibus https://identifiers.org/geo/GSE140696 (2019).
Jiang, L. et al. Synthetic spike-in standards for RNA-seq experiments. Genome res. 21, 1543–1551 (2011).
Brown, J., Pirrung, M. & McCue, L. A. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139 (2017).
Raymond, O. et al. The Rosa genome provides new insights into the domestication of modern roses. Nat genet. 50, 772–777 (2018).
Acknowledgements
This work is supported by National Natural Science Foundation of China (grants numbers 31772344 and 31972444) and The National Key Research and Development Program of China National Key Research and Development Project (2020YFD1000403) to ZZ.
Author information
Authors and Affiliations
Contributions
X.L., J.W. and F.J. designed and performed the experiments, de-novo assembly and functional annotations, submitted the data to Genbank and complemented the writing of the manuscript. X.C., Q.Z. and C.C. performed the experiments and contributed to data analysis and manuscript writing. N.M. and X.Z. planed the project, analyzed part of data and complemented the writing. Z.Z. planned the project, designed and supervised the experiments, coordinated the collaboration of the authors and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, X., Wu, J., Ji, F. et al. Transcriptomic profiling of rose flower under treatment of various phytohormones and plant growth regulators. Sci Data 9, 669 (2022). https://doi.org/10.1038/s41597-022-01800-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01800-w
