
Abstract
As part of a study identifying relationships between environmental variables and insect distributions within a bioenergy crop, giant miscanthus (Miscanthus x giganteus) samples were collected in October 2016 at 33 locations within a field in southeast Georgia, USA. At each location, one plant sample was collected every 3 to 4 m along a 15 m transect, resulting in 5 replicates per sampling location. The plant samples were separated into leaves and stems, dried, and ground. The chemical composition of the ground material was assessed by measuring total carbon and nitrogen, total macro- and micronutrients (aluminum, arsenic, boron, calcium, cadmium, cobalt, chromium, copper, iron, potassium, magnesium, manganese, molybdenum, sodium, nickel, phosphorus, lead, sulfur, selenium, silicon, titanium, vanadium, and zinc) using Inductively Coupled Plasma with Optical Emission Spectroscopy (ICP-OES), and optical characteristics of the water extractable organic matter (WEOM) using UV-Visible and Fluorescence Excitation Emission Matrix (EEM) spectroscopy. This dataset will be useful to identify relationships between the chemical composition of giant miscanthus tissues and pest distributions within a bioenergy crop field.
Measurement(s) | plant tissue chemical composition |
Technology Type(s) | inductively coupled plasma with optical emisison spectroscopy • UV-Visible and fluorescence spectroscopy |
Sample Characteristic - Organism | Miscanthus x giganteus |
Sample Characteristic - Environment | bioenergy crop field |
Sample Characteristic - Location | United States |
Similar content being viewed by others


Background & Summary
Miscanthus x giganteus (or giant miscanthus) is a perennial grass used increasingly as a bioenergy feedstock1. In the United States, the use of giant miscanthus has increased as producers seek productive biofeedstock alternatives to corn (Zea mays)2 that are more suitable for planting in marginal lands3. However, as a relatively new crop, we have only preliminary knowledge about insect pests of giant miscanthus, including the drivers of infestations and plant responses. Therefore, a study was undertaken in Georgia, USA, in 2015 and 2016 to ascertain more about the dynamics among insect pests, giant miscanthus plants, and broader field characteristics4. This work showed that wind speed, plant health, and soils are important drivers of phloem-feeding insect numbers but that these can vary over space and time. The dataset described here builds on an earlier dataset5 to provide detailed chemical characteristics of giant miscanthus plant tissues associated with the studies described by Coffin et al.3,4. These data will be useful to researchers focused on improving knowledge about the relationships among arthropod communities in giant miscanthus described earlier, and giant miscanthus plant tissue characteristics described here.
During a study conducted in 2016, giant miscanthus leaf and stem tissue samples were collected at 33 locations within a field, co-located with contemporaneous arthropod collection efforts (Fig. 1). Field management practices were previously described by Coffin et al.3,4. After completion of the arthropod collection campaign4, circular plots (r ≈ 8 m) centered at each sampling location were marked, and the crop was harvested from the surrounding areas. Aboveground plant tissue collection then occurred on 12 October 2016 along a 15 m transect centered on the undisturbed sampling locations, where one sample was collected every ~3.75 m, providing 5 sub-samples, or “reps” per circular plot.

Map of Miscanthus x giganteus data collection locations. Center panel – map of plant tissue sampling locations4. Upper left inset panel – general study location in TyTy, GA, denoted by a green star. Lower right inset panel – diagram of typical 15 m transects (red line centered, in gray, and extending across each circular plot), showing locations of sample collection points (black crosses) every ~3.75 m along the transect.
Methods
Geospatial data
Sampling locations were established, flagged, and recorded in June 2016, using a Trimble Geo7X global navigation satellite system (GNSS) receiver using the Trimble® VRS Now real-time kinematic (RTK) correction. Location accuracies were verified to within ±2 cm. Points were imported into a geodatabase using Esri ArcMap (Advanced license, Version 10.5) and projected using the Universal Transverse Mercator (UTM), Zone 17 North projection, with the 1983 North American datum (NAD83). Field investigators navigated to the flagged locations by visually locating them in the field or by using recreational grade GNSS receivers with the locations stored as waypoints.
Plant tissue sampling and preparation
Miscanthus x giganteus grows in clumps of bamboo-like canes. A single cane was cut at soil level from each of the five sample collection points in each circular plot, individually labelled, and brought to the lab for processing (Fig. 2). Each stem was measured from the cut at the base to the last leaf node, and the length was recorded. Green, fully expanded leaves were cut from each stem and leaves and stems from each plant were placed in separate paper bags and dried at 60 °C. The dry leaf and stem tissues were ground to pass a 1 mm screen (Wiley Mill Model 4, Thomas Scientific, Swedesboro, New Jersey, USA). Subsamples of the ground material were analyzed for total carbon (C) and nitrogen (N), acid-digested for the analysis of total macro- and micronutrients, and water-extracted for spectroscopic analysis and the characterization of the water extractable organic matter (WEOM) (Fig. 2).

Images of field samples, and diagram of plant tissue processing. Center panel – flow chart outlining the procedures for plant tissue processing, the kinds of analyses performed, and the type of data generated. Upper left inset panel – ground level picture of Miscanthus x giganteus circular plots. Upper right inset panel – some plant samples on the day of collection.
Total carbon and nitrogen
Dried and ground leaf and stem material (~4–6 mg) was analyzed for total C and N content by combustion (Vario EL III, Elementar Americas Inc., Mt. Laurel, New Jersey, USA). The instrument was calibrated using an aspartic acid standard (36.08% C ± 0.52% and 10.53% N ± 0.18%). Validation by inclusion of two aspartic acid samples as checks in each autosampler carousel (80 wells) resulted in a net positive bias of 1.44 and 1.68% for C and N, respectively. The mean C and N concentrations and standard deviations for the sample set are presented in Table 1.
Macro- and micronutrients
Plant tissue samples were analyzed for a suite of macro- and micronutrients including aluminum (Al), arsenic (As), boron (B), calcium (Ca), cadmium (Cd), cobalt (Co), chromium (Cr), copper (Cu), iron (Fe), potassium (K), magnesium (Mg), manganese (Mn), molybdenum (Mo), sodium (Na), nickel (Ni), phosphorus (P), lead (Pb), sulfur (S), selenium (Se), silicon (Si), titanium (Ti), vanadium (V), and zinc (Zn) using Inductively Coupled Plasma with Optical Emission Spectroscopy (ICP-OES). Samples (0.5 g) were digested using 10 mL of trace metal grade nitric acid (HNO3) in a microwave digestion system (Mars 6, CEM, Matthews, North Carolina, USA). During the digestion procedure (CEM Mars 6 Plant Material Method), the oven temperature was increased from room temperature to 200 °C in 15 minutes and held at 200 °C for 10 minutes. The pressure limit of the digestion vessels was set to 800 psi although it was not monitored during individual runs. Sample digestates were transferred quantitatively to centrifuge tubes, diluted to 50 mL with 2% HNO3 (prepared with lab grade deionized water), and centrifuged at 2500 rpm for 10 min (Sorvall ST8 centrifuge, Thermo Fisher Scientific, San Jose, California, USA). The digestates were decanted into clean centrifuge tubes and analyzed using an iCAP 7400 ICP-OES Duo equipped with a Charge Injection Device detector (Thermo Fisher Scientific, San Jose, California, USA). An aliquot of digested sample was aspirated from the centrifuge tube using a CETAC ASX-520 autosampler (Teledyne CETAC Technologies, Omaha, Nebraska, USA) and passed through a concentric tube nebulizer. The resulting aerosol was then swept through the plasma using argon as the carrier gas with a flow rate of 0.5 L/min and a nebulizer gas flow rate of 0.7 L/min. Macro- and micronutrients were quantified by monitoring the emission wavelengths (Em λ) reported in Table 2.
Characterization of the water extractable organic matter (WEOM)
The WEOM of the giant miscanthus leaves and stems was isolated by extracting the plant material with deionized water at room temperature6. The water extractions were performed by mixing ~0.2 g of dry, ground leaves and stems with 100 mL of deionized water in 125 mL pre-washed brown Nalgene bottles. All brown Nalgene bottles used for these extractions were pre-washed by soaking them for 24 hours in a 10% hydrochloric acid solution followed by 24 hours in a 10% sodium hydroxide solution, and a thorough rinse with deionized water. The bottles containing the extraction solution were shaken on an orbital shaker at 180 rpm for 24 hours. The extract was vacuum filtered using 0.45 µm glass fibre filters (GF/F, Whatman) into pre-washed 60 mL brown Nalgene bottles. The filtered water extracts containing the WEOM were stored in the dark in a refrigerator (4 °C) until analysis by UV-Visible and fluorescence spectroscopy. Samples were visually inspected just prior to analysis to ensure no colloids or precipitates had formed during storage. Samples that had become visually cloudy were re-filtered.
On the day of analysis, the water extracts were removed from the refrigerator and allowed to warm up to room temperature. Chemical characteristics of the WEOM were assessed through the analysis of optical properties on an Aqualog spectrofluorometer (Horiba Scientific, New Jersey, USA) equipped with a 150 W continuous output Xenon arc lamp. Excitation-emission matrix (EEM) scans were acquired in a 1 cm quartz cuvette with excitation wavelengths (Ex λ) scanned using a double-grating monochrometer from 240 to 621 nm at 3 nm intervals. Emission wavelengths (Em λ) were scanned from 246 to 693 nm at 2 nm intervals and emission spectra were collected using a Charge Coupled Device (CCD) detector. All fluorescence spectra were acquired in sample over reference ratio mode to account for potential fluctuations and wavelength dependency of the excitation lamp output. Samples were corrected for the inner filter effect7 and each sample EEM underwent spectral subtraction with a deionized water blank to remove the effects due to Raman scattering. Rayleigh masking was applied to remove the signal intensities for both the first and second order Rayleigh lines. Instrument bias related to wavelength-dependent efficiencies of the specific instrument’s optical components (gratings, mirrors, etc.) was automatically corrected by the Aqualog software after each spectral acquisition. The fluorescence intensities were normalized to the area under the water Raman peak collected on each day of analysis and are expressed in Raman-normalized intensity units (RU). All sample EEM processing was performed with the Aqualog software (version 4.0.0.86).
The optical data obtained from the EEM scans were used to calculate several indices representative of WEOM chemical composition (Table 3) including the absorbance at 254 nm (Abs254), the ratio of the absorbance at 254 to 365 nm (Abs254:365), the ratio of the absorbance at 280 to 465 nm (Abs280:465), the spectral slope ratio (SR), the fluorescence index (FI), the humification index (HIX), the biological index (BIX), and the freshness index (β:α). The SR was calculated as the ratio of two spectral slope regions of the absorbance spectra (275–295 and 350–400 nm)8. The FI was calculated as the ratio of the emission intensities at Em λ 470 and 520 nm, at an Ex λ of 370 nm9. The HIX was calculated by dividing the emission intensity in the 435–480 nm region by the sum of emission intensities in the 300–345 and 435–480 nm regions, at an Ex λ of 255 nm10. The BIX was calculated as the ratio of emission intensities at 380 and 430 nm, at an Ex λ of 310 nm11. The freshness index β:α was calculated as the emission intensity at 380 nm divided by the maximum emission intensity between 420 and 432 nm, at an Ex λ of 310 nm12. To further characterize the giant miscanthus WEOM, the fluorescence intensity at specific excitation-emission pairs was also identified. The fluorescence peaks identified here have previously been reported for surface water samples and water extracts13 and include peak A (Ex λ 260, Em λ 450), peak C (Ex λ 340, Em λ 440), peak M (Ex λ 300, Em λ 390), peak B (Ex λ 275, Em λ 310), and peak T (Ex λ 275, Em λ 340). A brief description of these optical indices is provided in Table 3.
Data Records
The data records include measurements for giant miscanthus leaf and stem chemical composition and WEOM characterization. Each data record includes identifying information regarding its sample number, circular plot location, replicate number, latitude and longitude coordinates, and whether the sample was taken from leaf or stem tissues. Chemical composition measurements include the total macro- and micronutrients described above, with corresponding MDL values, nutrient concentrations with MDL values shown where true values were less than the MDL, and true concentration values, for each nutrient. Dry weight is provided for each sample, and C and N concentrations are provided as a percentage of dry weight. For stems, length is provided. Spectroscopic measurements follow for each record, including the absorbance at 254 nm (Abs254), the ratios of the absorbance at 254 and 365 nm (Abs254:365) and at 280 and 465 nm (Abs280:465), the spectral slope ratio (SR), the fluorescence index (FI), the humification index (HIX), the biological index (BIX), the freshness index (β:α), and the fluorescence intensity peaks A, C, M, B, and T.
Data records are archived in the USDA National Agricultural Library, Ag Data Commons repository and are publicly available14. Data files include identical versions of the data in two formats: a .xlsx file and a .xml file. A data dictionary.txt file provides metadata for the.xml version, while an identical metadata tab is included in the .xlsx.
The raw spectroscopic data are available as .dat files from the corresponding author upon request.
Technical Validation
Digestion procedures and ICP analysis
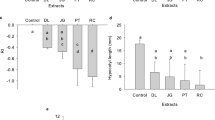
All digestion vessels were cleaned between batches with soap and water followed by the CEM Xpress Clean method and a thorough deionized water rinse. To establish digestion proficiency and baseline performance of the microwave digestion system, a Soybean Meal Certified Reference Material (CRM-SBM-S, High Purity Standards, North Charleston, South Carolina, USA) was digested in duplicate with each batch (40 samples per batch) and the average percent recovery for all macro- and micronutrients was calculated. All nutrients were recovered with less than 10% error and the percent recoveries ranged from 95% for Al to 109% for Ca, Fe, Mg, and Mo. To assess and visualize the accuracy of the digestion and ICP analyses, a comparison was made between the true, certified nutrient concentrations of the Soybean Meal CRM (reported in the certificate of analysis) and the measured CRM nutrient concentrations (Fig. 3). The low standard deviations observed between the true and measured concentrations indicate the accuracy and precision of the digestion and ICP procedures.

Comparison between the true and the measured nutrient concentrations of the Soybean Meal CRM. Left panel – entire concentration range for the nutrients quantified. Right panel - an expansion showing the low concentration range of the nutrients quantified.
All the standards used for calibration of the ICP were prepared gravimetrically and nutrient concentrations were determined in mg/kg (ppm). A calibration curve was run at the beginning of each ICP sequence with calibration ranges from 0.001 mg/kg to 100 mg/kg. Because of variable concentrations in the samples, some nutrients required low (S, Se, Si, and V) and high calibration points (B, Cd, Co, Cr, Mo, Ni, Pb, Se, Ti, V, and Zn). Samples were run with 1 and 10 ppm calibration verification standards that were added to the sequence to monitor the stability and accuracy of the calibration. Duplicate analyses of the Soybean Meal CRM were run with each instrument sequence to monitor accuracy and precision.
A blank-based MDL was calculated as the mean blank plus t times the standard deviation of all the blanks digested and analysed for this study. The greater of the blank-based MDL and the low calibration point was multiplied by the dilution factor for each sample to generate accurate, sample-specific detection limits for each analyte.
Spectroscopic data
Although optical measurements are simple and straightforward, they can be affected by many variables such as inconsistencies in sample preparation (e.g., filter pore size, sample storage and preservation), in the correction of interferences (e.g., sample dilution, inner filter corrections), and in dealing with instrument drift (lamp intensity). The optical dataset reported here was obtained by following a documented, standardized procedure for the operation of the Aqualog spectrofluorometer to ensure that the optical measurements collected are useful and widely comparable15. On each day of sample analysis, the instrument was warmed up for 1 hour prior to the first scan. Lamp hours were recorded as soon as the instrument was turned on. Because the lamp degrades over time, it is routinely replaced annually regardless of hours of operation (the manufacturer recommends changing the lamp after 1000 hours of use). Gloves were always worn when handling the cuvettes and samples. All laboratory blanks and sample dilutions were prepared with deionized water (18.2 MΩ resistance, ≤ 5 ppb total organic C) produced with a Milli-Q Advantage A10 water purification system (Millipore Sigma, Burlington, Massachusetts, USA). To avoid sample contamination due to bacterial breakthrough from resin beds, activated C, and filters, the water purification system used in the lab was maintained and monitored frequently for background C and bacterial growth. The cuvette was rinsed between each sample with plenty of deionized water.
To track and maintain instrument performance, two validation experiments were run on each day of analysis: The excitation validation scan and the water Raman signal-to-noise ratio and emission calibration scan. The excitation validation scan was used to verify lamp performance and peak position at 467 ± 1 nm. The water Raman signal-to-noise and emission calibration scan examines the wavelength calibration of the CCD detector and consists in the emission scan of the Raman-scatter band of water15. This scan was used to verify the peak location of the water Raman at 397 ± 1 nm and the Raman peak area was used to normalize the fluorescence signals, producing data that can be compared across instruments. Multiple laboratory blanks (6–9) were analysed on each day of analysis to obtain the signal associated with the instrument, cuvette, and deionized water in the absence of fluorescent WEOM. The daily blanks were subtracted from the analytical set run on the same day.
Optical measurements of highly concentrated samples can be subject to measurement errors such as inner filter effects7. The inner filter effect has been found to be linear and correctable for natural water samples when the Abs254 is between 0.03–0.3 absorbance units (AU) when measured in a 1 cm cuvette10,16. To standardize the correction procedure used for this dataset, the WEOM samples were initially analysed at full concentration and the Abs254 was recorded. If the Abs254 exceeded 0.3 AU, the sample was diluted to a concentration at which the Abs254 was in the range of 0.3–0.3 AU and corrections for the inner filter effect were applied.
Usage Notes
Pertaining to the macro- and micronutrient data: To ascertain which concentration records are lower than the corresponding MDL, a comparison of the XX_Conc_MDL and XX_Conc_True fields can be done, where affected records are true when XX_Conc_True is less than XX_Conc_MDL (XX is the element name).
The data are ready for use with a geographic information system (GIS), where longitude and latitude coordinates are projected as noted above. Since exact sample point locations along transects were not measured in the field, users are advised to consider the replicate values as pertaining to the area of the circular plot (r ≈ 8 m) at the single sampling location provided by the coordinates.
Code availability
No custom code was used for the generation or analysis of this data.
References
Arnoult, S. & Brancourt-hulmel, M. A Review on Miscanthus Biomass Production and Composition for Bioenergy Use: Genotypic and Environmental Variability and Implications for Breeding. BioEnergy Research 8, 502–526, https://doi.org/10.1007/s12155-014-9499-4:1-11 (2015).
Mitchell, R. B. et al. Dedicated Energy Crops and Crop Residues for Bioenergy Feedstocks in the Central and Eastern USA. BioEnergy Research 9, 384–398, https://doi.org/10.1007/s12155-016-9734-2 (2016).
Coffin, A. et al. Potential for Production of Perennial Biofuel Feedstocks in Conservation Buffers on the Coastal Plain of Georgia, USA. BioEnergy Research, 1–14, https://doi.org/10.1007/s12155-015-9700-4 (2015).
Coffin, A. W. et al. Responses to environmental variability by herbivorous insects and their natural enemies within a bioenergy crop, Miscanthus x giganteus. PLOS ONE 16, e0246855, https://doi.org/10.1371/journal.pone.0246855 (2021).
Coffin, A. W. Data from: Responses to environmental variability by herbivorous insects and their natural enemies within a bioenergy crop, Miscanthus x giganteus. Ag Data Commons https://doi.org/10.15482/USDA.ADC/1520060 (2020).
Monda, H., Cozzolino, V., Vinci, G., Spaccini, R. & Piccolo, A. Molecular characteristics of water-extractable organic matter from different composted biomasses and their effects on seed germination and early growth of maize. Science of The Total Environment 590-591, 40–49, https://doi.org/10.1016/j.scitotenv.2017.03.026 (2017).
McKnight, D. M. et al. Spectrofluorometric characterization of dissolved organic matter for indication of precursor organic material and aromaticity. Limnology and Oceanography 46, 38–48, https://doi.org/10.4319/lo.2001.46.1.0038 (2001).
Helms, J. R. et al. Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter. Limnology and Oceanography 53, 955–969, https://doi.org/10.4319/lo.2008.53.3.0955 (2008).
Cory, R. M. & McKnight, D. M. Fluorescence Spectroscopy Reveals Ubiquitous Presence of Oxidized and Reduced Quinones in Dissolved Organic Matter. Environmental Science & Technology 39, 8142–8149, https://doi.org/10.1021/es0506962 (2005).
Ohno, T. Fluorescence Inner-Filtering Correction for Determining the Humification Index of Dissolved Organic Matter. Environmental Science & Technology 36, 742–746, https://doi.org/10.1021/es0155276 (2002).
Huguet, A. et al. Properties of fluorescent dissolved organic matter in the Gironde Estuary. Organic Geochemistry 40, 706–719, https://doi.org/10.1016/j.orggeochem.2009.03.002 (2009).
Parlanti, E., Wörz, K., Geoffroy, L. & Lamotte, M. Dissolved organic matter fluorescence spectroscopy as a tool to estimate biological activity in a coastal zone submitted to anthropogenic inputs. Organic Geochemistry 31, 1765–1781, https://doi.org/10.1016/S0146-6380(00)00124-8 (2000).
Coble, P. G. Characterization of marine and terrestrial DOM in seawater using excitation-emission matrix spectroscopy. Marine Chemistry 51, 325–346, https://doi.org/10.1016/0304-4203(95)00062-3 (1996).
Pisani, O., Liebert, D., Strickland, T. C. & Coffin, A. W. Data from: Plant Tissue Characteristics of Miscanthus x giganteus. Ag Data Commons https://doi.org/10.15482/USDA.ADC/1524724 (2022).
Hansen, A. M. et al. Procedures for using the Horiba Scientific Aqualog® fluorometer to measure absorbance and fluorescence from dissolved organic matter. Report No. 2018-1096, (Reston, VA, 2018).
Miller, M. P. et al. New light on a dark subject: comment. Aquatic Sciences 72, 269–275, https://doi.org/10.1007/s00027-010-0130-2 (2010).
Acknowledgements
This research was funded by the United States Department of Agriculture-Agricultural Research Service (USDA-ARS), National Program 211, Water Availability and Watershed Management, Project #6048-13000-026-00D. This research is a contribution from the Long-Term Agroecosystem Research (LTAR) network, supported by the USDA. The authors are grateful for the assistance of the following research technicians with the USDA-ARS Southeast Watershed Research Laboratory: Sally Belflower, Rex Blanchett, Lorine Lewis, Josie McCully, Josh Moore, Coby Smith, and Margie Whittle.
Author information
Authors and Affiliations
Contributions
O.P., T.C.S. and A.W.C. conceptualized and designed the research; O.P., D.L., T.C.S. and A.W.C. performed the experiments and collected the data; O.P., D.L., T.C.S. and A.W.C. processed, summarized, and curated the data; O.P., D.L., T.C.S. and A.W.C. wrote the initial draft of the manuscript; O.P. and A.W.C. reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pisani, O., Liebert, D., Strickland, T.C. et al. Plant tissue characteristics of Miscanthus x giganteus. Sci Data 9, 308 (2022). https://doi.org/10.1038/s41597-022-01424-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01424-0
