
Abstract
The generation of omic resources is central to develop adequate management strategies for species with economic value. Here, we provide high-coverage RNA-seq datasets of liver tissue (containing between 80,2 and 88,4 million of paired-end reads) from four wildtype teleost species with high commercial value: Trachurus trachurus (TTR; Atlantic horse mackerel), Scomber scombrus (SSC; Atlantic mackerel), Trisopterus luscus (TLU; pout), and Micromesistius poutassou (MPO; blue whiting). A comprehensive assembly pipeline, using de novo single and multi-kmer assembly approaches, produced 64 single high-quality liver transcriptomes – 16 per species. The final assemblies, with N50 values ranging from 2,543–3,700 bp and BUSCO (Benchmarking Universal Single-Copy Orthologs) completeness values between 81.8–86.5% of the Actinopterygii gene set, were subjected to open reading frame (ORF) prediction and functional annotation. Our study provides the first transcriptomic resources for these species and offers valuable tools to evaluate both neutral and selected genetic variation among populations, and to identify candidate genes for environmental adaptation assisting in the investigation of the effects of global changes in fisheries.
Measurement(s) | RNA • transcriptome • sequence_assembly • sequence feature annotation |
Technology Type(s) | RNA sequencing • sequence assembly process • sequence annotation |
Factor Type(s) | teleost species |
Sample Characteristic - Organism | Trachurus trachurus • Scomber scombrus • Trisopterus luscus • Micromesistius poutassou |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12533171
Similar content being viewed by others
Background & Summary
Multi-data approaches using complementary techniques are essential to successfully define fish stocks and management strategies (e.g.1,2). The revolution of Next-Generation Sequencing (NGS) has created an unprecedented opportunity to contribute to each component of fisheries management (e.g. reviewed in3), allowing to address population structure and adaptive divergence in commercially relevant teleost fish species (e.g. Atlantic cod4 and Atlantic herring5), to identify candidate genes for environmental adaptation3 or to explore the function of genes with aquaculture relevance6. Importantly, the expansion of genomic and transcriptomic datasets has been fundamental to detail the complex phylogenetic relationships of this taxon-rich clade7,8,9. Additionally, these resources have also been proven important in conservation strategies, where they have allowed the prediction of how species will respond to new environmental scenarios and the identification of the threats endangering species at risk (e.g.10). Yet, these formidable tools have to be applied to the vast majority of the world fisheries: 60% of which are at their maximum sustainable yield, 33% are exploited at biologically unsustainable levels and only 7.0% are considered underfished11.
Here, we generated four liver transcriptomic datasets from important fishing resources in European waters belonging to three different taxonomic families: the Atlantic horse mackerel, Trachurus trachurus (Linnaeus 1758), the Atlantic mackerel, Scomber scombrus (Linnaeus 1758) (SSC), the pout, Trisopterus luscus (Linnaeus 1758) and the blue whiting, Micromesistius poutassou (Risso 1827) (Fig. 1a). Together, these species represent an important fraction of fish captures in European waters (Fig. 1a) and are, therefore, relevant models for which the development of omic tools for research is highly desirable. Although some information regarding basic biological traits is currently available, some aspects of their biology remain poorly known, particularly population structure, nutritional requirements, reproduction, coastal recruitment processes, and studies involving NGS datasets are also scarce or absent. For example, in Scomber scombrus two RNA-seq projects are available (PRJNA272777 (low coverage RNA-seq dataset produced from a pool of tissues (muscle, liver, gonad, brain) and 454 GS-FLX Titanium System) and PRJNA305977 (RNA-seq dataset produced from white muscle tissue and with Illumina HiSeq2000 system))12,13. For the remaining species, no RNA-seq data is currently available. To cover the above-mentioned biological aspects, the liver was selected to produce a high-quality and coverage transcriptome for each species. Tipically, this tissue has a large number of expressed genes. On the other hand, the selection of a single specimen per species avoided the intraspecies variations, which associated with heterozygosity levels of marine fish species, generally higher than freshwater species, makes the construction of high-quality genomic and transcriptomic references a complex and challenging task.

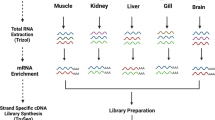
Schematic overview of the study. (a) Geographic coordinates of sampling, photographs of the collected specimens, taxonomy classification and fishery relevance. (b) Experimental setup used to perform RNA extraction and sequencing. (c) Bioinformatics workflow used to post-sequencing dataset analyses.
Methods
Animal sampling
Adult specimens of T. trachurus, S. scombrus, T. luscus, and M. poutassou were collected to perform the RNA-seq analyses. Individuals were caught in the Northeast Atlantic Ocean, Portugal: T. trachurus and S. scombrus (40.961667 N, 9.336000 W) and T. luscus and M. poutassou (41.055000 N, 9.246667 W), under the guidelines of the “Programa Nacional de Amostragem Biológica”, conducted by the Instituto Português do Mar e da Atmosfera (IPMA) (Fig. 1a; Table 1). Immediately upon capture, liver tissue from each specimen was collected and stored in RNAlater RNA Stabilization Reagent (Qiagen, Germany) during 24 h at 4 °C (Fig. 1b). The samples were later transferred to −80 °C until total RNA extraction (Fig. 1b).
RNA extraction, library construction, and sequencing
Total RNA was extracted from liver using the Illustra RNAspin Mini RNA Isolation Kit (GE Healthcare, UK), according to the manufacturer’s instructions. The isolated RNA was treated with RNase-free DNase I to remove residual genomic DNA contamination and eluted in RNase-free water. RNA concentration was measured using a microplate spectrophotometer with Take3™ Micro-Volume Plate (BioTeK, USA) (T. trachurus - 2816,556 ng/μl, S. scombrus - 2379,382 ng/μl, T. luscus - 1147.368 ng/μl, and M. poutassou - 1236.980 ng/μl). The RNA quality was verified with the measurement of the OD260/280 ratio values (1.8 to 2.0). The integrity of each RNA sample was checked by running 1 μl in a 1% agarose gel. Afterwards, the four RNA samples were used to build four strand-specific libraries, one per species, with an insert size of 250–300 bp and sequenced using 150 bp paired-end reads on the Illumina HiSeq4000 platform by Novogene (China).
Pre-assembly processing stage
The raw dataset for each specimen was initially inspected with the FastQC (version 0.11.8) software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic (version 0.38)14 was then used to trim, quality-filter the raw reads and remove Illumina adaptors, under the following parameters (LEADING:5 TRAILING:5 SLIDINGWINDOW:5:20 MINLEN:36) (Fig. 1c). To correct random sequencing errors introduced during the sequencing or in another stage of the pre-in silico processing, we applied a kmer-based error correction method, Rcorrector (version 1.0.3)15, with default settings. At this stage, all the unfixable reads were discarded. The error-corrected reads were posteriorly introduced in the Centrifuge (version 1.0.3-beta)16 program and taxonomically classified against the pre-compiled nucleotide database of NCBI (ftp://ftp.ccb.jhu.edu/pub/infphilo/centrifuge/data/) (version nt_2018_3_3). Importantly, all the reads not classified as belonging to Actinopterygii superclass (Taxon Id: 7898) were considered exogenous to our target species and removed from the initial datasets (Fig. 1c).
De novo transcriptome assembly stage
To build the transcriptome, we opted by the de novo assembly strategy using a multi-kmer approach. Thus, to generate the liver transcriptomes of TTR, SSC, TLU, and MPO we used four assemblers – Trinity (version 2.8.4)17,18, rnaSPAdes mode of SPAdes (version 3.11.1)19, SOAPdenovo-trans (version 1.03)20, and IDBA-trans (version 1.1.3)21 (Fig. 1c). The first assembly was carried out by Trinity software with a fixed Kmer of 25 and a strand-specific data parameter on (SS_lib_type RF). The RnaSPAdes tool was then applied with the default parameters (kmer of 73, following the strategy used in the original publication (read length/2−1)19. In the remaining assemblers, we used a multi-kmer approach that required both the mean insert size (IS) and standard deviation (SD) values of the raw dataset. To calculate these values, we used the transcriptome generated by Trinity as a reference, the Bowtie2 (version 2.3.5)22 to map the clean raw reads, and finally CollectInsertSizeMetrics function of Picard tools (version 2.19.2)23 to estimate the insert size and standard deviation values. The IDBA-trans assemblies were performed with the SD and IS, previously calculated, the kmer values of 25, 31, 41, 51, 61, 71, 81 and the–ss-fr parameter on. On the other hand, SOAPdenovo-trans used the kmer values of 25, 31, 41, 51, 61, 71, 81 with (-L 200; -F YES) parameters. To remove the gaps inserted during the SOAPdenovo-trans assemblies, the GapCloser (version 1.12) module of SOAPdeonovo224 software was used, with default settings.
Post-assembly processing stage
In the post-assembly processing stage, all assemblies were processed with the SeqKit (version 0.10.1) toolkit25. This tool removed all contigs with less than 200 nucleotides, and concatenated all assemblies, per species, in one multi-assembly file. Furthermore, we also conducted several searches against the nucleotide NCBI (nt-NCBI) (downloaded on 30/03/2019) and UniVec (downloaded on 02/04/2019) databases to identify and remove biological contaminations, vectors or adapters not identified in the previous stages. These searches were done via blast-n (version 2.9.0) against the nt-NCBI database with the parameters (-evalue 1e-5; -max_target_seqs. 1; -perc_identity 90; -max_hsps 1; and minimum alignment length of 50 bp), and against UniVec database with the settings (-reward 1; -penalty -5; -gapopen 3; -gapextend 3; -dust yes; -soft_masking true; -evalue 700; -searchsp 1750000000000). For the nt-NCBI searches, all contigs with the best match hits out of the Actinopterygii taxon were considered contaminations and removed from the transcriptome assemblies. The remaining transcripts, without any match hit or with match hits in Actinopterygii taxon were kept into the transcriptomes and used for further analyses. Regarding the Univec database, all transcripts with a match hit were considered exogenous and removed from the dataset. To decrease the redundancy and complexity within the decontaminated transcriptomes, we clustered highly similar nucleotide sequences with the CD-HIT-EST (version 4.7)26 software, with the following settings (-c 0.95; -g 1; -M 60000; -T 30) (Fig. 1c). Essentially, the software clusters and compares nucleotide sequences, keeping the longest sequence per cluster above a certain similarity threshold, in our case 95% of similarity. All steps of the transcriptome assembly and post-processing stage (from the single kmer assemblies build until the concatenation, decontamination and the clean-up of redundancy) were further inspected to guarantee the accuracy of our approach. Thus, we used the Trinity and Transrate (1.0.3)27 for primary statistics and the Benchmarking Universal Single‐Copy Orthologs (BUSCO version 3.0.2)28 – with four lineage-specific profile libraries (Metazoa, Eukaryota, Vertebrates and Actinopterygii) – to evaluate the gene completeness of each assembly. In addition, the rate of reads back mapping to the transcriptome (RBMT) was also calculated for all the assemblies after the decontamination step (Fig. 1c). The RBMT was performed with Bowtie2 (version 2.3.5)22 tool.
Open reading frame prediction and transcriptome annotation
The open reading frames (ORFs) were predicted using the Transdecoder software (version 5.3.0) (https://transdecoder.github.io/) (Fig. 1c). This pipeline is mainly subdivided into three stages. In the first stage, the software pre-predicted the longest ORF per transcript with a cut off length of 100 aminoacids. In the second stage, to find homology and protein evidence, two databases were screened – blast-p (version 2.9.0) with cut-off evalue of 1e-5 against UniProtKB/Swiss-Prot database (downloaded on 12/04/2019)29 and hmmscan of hmmer2 package (version 2.4i)30 to find protein profiles against PFAM database (downloaded on 12/04/2019)31, respectively. In the last stage, all the information collected from both databases, together with the pre-predicted ORF’s were used to perform the final prediction of the amino acid sequence. Afterwards, all transcripts codifying for a protein, per species, were used to carry out a functional annotation step with the Sma3s (version 2.1)32 tool (Fig. 1c). Functional annotation was assigned applying consecutive filters to a blast record (performed against the Uniref90 database (downloaded on 2019-02)) based on both similarity and query coverage. Functional domains were also identified clustering all significant blast hits, and their annotations were retrieved only when their frequency was higher than the frequency of appearance in the reference database following the hypergeometric distribution. Annotation types retrieved consist of GO terms, EC codes from ENZYME repository and Uniprot keywords and pathways. Gene name is only associated in cases of a blast hit greater than 75% of identity and 75% of query coverage.
Data Records
The data generated in this study is subdivided in three main categories: the raw reads, the transcriptome assemblies, and the functional annotation. The decontaminated raw reads, for each species, were deposited in the NCBI Sequence Read Archive – SRP21618733. Furthermore, the non-redundant transcriptome assemblies were submitted in fasta format, to the NCBI Transcriptome Shotgun Assembly Sequence Database, under the accession number, GHRS00000000, (TTR), GHRT00000000 (SSC), GHRZ00000000 (TLU), GHRY00000000 (MPO)34,35,36,37. The remaining transcriptome assemblies (fasta format), the open reading frames, as well the annotation files per species were uploaded to figshare online repository38. In detail, it is possible to consult in figshare, the 16 initial individual transcriptome assemblies of the four species produced with four different assemblers, the multi assembly and the final assembly, per species, in fasta format. In addition, also the predicted open reading frames (.pep file), as well as the remaining outputs of TransDecoder software (.bed,.gff3 and.cds files) and the functional annotation files are available, per species38.
Technical Validation
Raw datasets and pre-assembly processing quality control
The sequencing process generated a total of 88,4 M in TTR, 87,8 M in SSC, 80,2 M in TLU and 84 M in MPO paired-end raw reads. All raw datasets were initially scrutinized by the FastQC tool, trimmed with Trimmomatic, error-corrected with Rcorrector and decontaminated with Centrifuge software. The percentage of removed reads per step in the pre-assembly processing stage can be visualized in Table 2. Overall, ~86,7 M in TTR, 87,1 in SSC, 79,2 in TLU and 82,8 MPO reads had Phred scores higher than 20 and were retained for the transcriptome assembly stage (Table 2 and Fig. 2a–d33).

Quality FastQC report of post-processing RNA-seq datasets (after Centrifuge (version 1.0.3-beta)16 cleaning stage). For each species, it is presented the R1 and R2 FastQC reports. (a) Trachurus trachurus; (b) Scomber scombrus; (c) Trisopterus luscu; (d) Micromesistius poutassou.
Transcriptome assembly metrics
The de novo transcriptome assembly was performed using multiple software including Trinity, rnaSPAdes, SOAPdenovo-trans and IDBA-trans. This strategy has been previously applied with success in transcriptome projects of multiple organisms, without a reference genome39,40,41. The first assembly obtained with Trinity tool was used to map the raw decontaminated reads and to calculate the IS and SD for each species. In all species, the IS and SD values ranged between 267–280 and 59–61 (Table 2). The initial multi-assembly approach generated 64 transcriptome assemblies, 16 for each species38. The statistic metrics for each assembly, such as N50 transcript length, mean transcript length, percentage of GC, longest transcript length, or transcript number over 1k or 10k nucleotides can be visualized in Online-only Tables 1–4. In addition, we also performed BUSCO analyses using 303, 978, 2586 and 4584 near-universal single-copy orthologs in Eukaryota, Metazoa, Vertebrata, and Actinopterygii gene sets, respectively, for all the 16 assemblies (Online-only Tables 1–4). Regarding the individual assemblies, the Trinity tool presented the higher values of N50 and mean transcript length in all species: TTR – 1708 and 921,55 bp, SSC – 1964 and 984,48 bp, TLU – 1321 and 780,05 bp, and MPO – 1228 and 782.03 bp, respectively (Online-only Tables 1–4). Moreover, the BUSCO analyses revealed the same pattern with higher percentage of total genes found (complete + fragmented) in Trinity assemblies for all lineage-specific profile libraries consulted (Eukaryota, Metazoa, Vertebrata and Actinopterygii): TTR – 100.00, 99.28, 90.37, 81.72%, SSC – 99.01, 99.28, 89.29, 79.89%, TLU – 100.00, 99.80, 92.34, 83.40%, and MPO – 100.00, 99.69, 92.42, 83.99% (Online-only Tables 1–4). The magnitude of these values is comparable and, in some cases, superior to several fish transcriptomes, publicly available, e.g. (e.g. Xiphias gladius – 99 and 98.2% of genes found in Eukaryota, Metazoa databases42; Amphiprion percula – 85.4% of genes found in Actinopterygii database43), which suggests a high quality of our initial assemblies.
Post-assembly processing and annotation verification
At this stage, the 16 assemblies per species were concatenated and decontaminated, resulting in four multi-assembly transcriptomes – Online-only Table 438. During the decontamination phase, all blast-n results were manually inspected and the threshold of minimum alignment length of 50 bp and taxonomic superclass Actinopterygii, were specifically selected due to two factors: the considerable number of plausible biological contamination hits with more 50 bp of nucleotide alignment length (e.g. Lasthenia californica (Taxon ID: 149440)) and the relatively high number of Actinopterygii species with genome sequenced and annotated on nt-NCBI database (at least 43 species), respectively. Although, some transcripts have been discarded at this stage (e.g. novel sequences not present in this 43 Actinopterygii species), we have ensured the application of sensitive blast parameters. Thus, instead of focusing the analyses on the removal of all sequences with a match hit, we performed first an effort on the identification of the hits, via homology, against the possible sources of contamination and only after excluded if confirmed. Apart from a few transcripts that have been removed, this approach increased the confidence levels of each dataset. Posteriorly, four multi-assemblies were subject to a redundancy removal step to decrease the complexity and to remove the overlapping transcripts above 95% of sequence similarity34,35,36,37. This method has been highly used to remove redundancy in several datasets and organisms40,44,45,46, namely in the build of new transcriptomic references. In addition, this strategy still decreased the natural heterozygoty of the organisms in the assemblies. It should be noted that although naturally present in the organisms, the heterozygosity continues to be a crucial parameter to consider during the generation of transcriptomic and genomic datasets. In some cases, where heterozygosity is not contemplated in the bioinformatic approach, the results change completely and their interpretation can be affected. Using this approach, the total number of transcripts in each dataset was substantially reduced, TTR – 2769441 to 414729, SSC – 2728965 to 377586, TLU – 3203445 to 548983, and MPO – 3675167 to 602418, allowing a better overview and understanding of the datasets (Online-only Table 5). After this step, we implemented another quality control strategy, the RBMT. This method, together with the already established metrics, general statistics, and gene completeness, allowed us to verify the raw read content of the transcriptomes. Importantly, this metric showed that even with the redundancy removal step, the RBMT rate in the non-redundant transcriptomes has kept very high values, TTR – 96.78, SSC – 97.16, TLU – 95.3, MPO – 95.51% (Online-only Table 5).
In the process of ORF prediction, the TransDecoder software identified TTR – 111866, SSC – 97811, TLU – 150334, MPO – 167124 transcripts with an assigned ORF. Importantly, the entire set of transcripts codifying for a protein was collected from the non-redundant transcriptomes and placed in files classified as final transcriptome assemblies38.
The basic metrics, BUSCO analyses and RBMT rates for TTR, SSC, TLU, and MPO final transcriptome assemblies are available in Table 3. Notwithstanding, the final transcriptome statistics demonstrate the power of the assembly and processing strategy chosen, with N50 values ranging from 2543 to 3700 bp, BUSCO values between 81.8–86.5% in Actinopterygii gene set, and raw reads rate mapping ranging from 91.45 to 94.63% (Table 3). Interestingly, the BUSCO analyses in Vertebrata and Actinopterygii gene sets still shown a decrease in the percentage of fragmented genes (less than half) and missing genes (slightly), in the final transcriptome assemblies when compared with the initial individual assemblies, for all species.
In the end, the final transcriptomes were functionally annotated using the Sma3s software. A high number of the transcripts were annotated and most of them including the gene name which suggests a remarkable quality of the assemblies. Annotation distribution across the different species is very similar, keeping a logical proportion based on the total number of transcripts. All the annotations stats, including the gene homology, the most probable gene name, the GO terms, Kegg Pathways and EC numbers for Enzymes can be consulted in Table 338.
Code availability
All the software programs used in the bioinformatics workflow (transcriptome assembly, pre and post-assembly processing stages and transcriptome annotation) are presented in the Methods section. All programs and databases have the versions, download dates, and parameters described. Software programs with no parameters associated were used with the default settings.
References
Begg, G. A. & Waldman, J. R. An holistic approach to fish stock identification. In Fisheries Research 43, 35–44 (Elsevier, 1999).
Cadrin, S. X., Kerr, L. A. & Mariani, S. Stock Identification Methods: An Overview. In Stock Identification Methods: Applications in Fishery Science: Second Edition 1–5 (Elsevier Inc., 2013).
Kelley, J. L., Brown, A. P., Therkildsen, N. O. & Foote, A. D. The life aquatic: advances in marine vertebrate genomics. Nat. Rev. Genet. 17, 523–534 (2016).
LIMBORG, M. T. et al. Environmental selection on transcriptome-derived SNPs in a high gene flow marine fish, the Atlantic herring (Clupea harengus). Mol. Ecol. 21, 3686–3703 (2012).
Lamichhaney, S. et al. Population-scale sequencing reveals genetic differentiation due to local adaptation in Atlantic herring. Proc. Natl. Acad. Sci. U. S. A. 109, 19345–50 (2012).
Machado, A. et al. Out of the Can: A Draft Genome Assembly, Liver Transcriptome, and Nutrigenomics of the European Sardine, Sardina pilchardus. Genes (Basel). 9, 485 (2018).
Ravi, V. & Venkatesh, B. The divergent genomes of teleosts. Annu. Rev. Anim. Biosci. 6, 47–68 (2018).
Hughes, L. C. et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl. Acad. Sci. U. S. A. 115, 6249–6254 (2018).
Sun, Y. et al. Fish-T1K (Transcriptomes of 1,000 Fishes) Project: large-scale transcriptome data for fish evolution studies. Gigascience 5, 18 (2016).
Gonzalez, E. G. et al. Population Proteomics of the European Hake (Merluccius merluccius). J. Proteome Res. 9, 6392–6404 (2010).
FAO. State of world fisheries and aquaculture (SOFIA) - Meeting the sustainable development goals. Food and Agriculture Organization of the United Nations (2018).
Álvarez, P. et al. Genomic Resources Notes Accepted 1 June 2015–31 July 2015. Mol. Ecol. Resour. 15, 1510–1512 (2015).
Ciezarek, A. G. et al. Substitutions in the glycogenin-1 gene are associated with the evolution of endothermy in sharks and tunas. Genome Biol. Evol. 8, 3011–3021 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Song, L. & Florea, L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 4, 48 (2015).
Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8 (2013).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 8 (2019).
Xie, Y. et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666 (2014).
Peng, Y. et al. IDBA-tran: a more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 29, i326–i334 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–303 (2010).
Luo, R. et al. Erratum to ‘SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler’ [GigaScience, (2012), 1, 18]. Gigascience 4, 1 (2015).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11, e0163962 (2016).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Smith-Unna, R. D. et al. TransRate: reference free quality assessment of de novo transcriptome assemblies. Genome Res. 26, 1134–44 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Bateman, A. et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–301 (2012).
Casimiro-Soriguer, C. S., Muñoz-Mérida, A. & Pérez-Pulido, A. J. Sma3s: A universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics 17, 1700071 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP216187 (2020).
Machado, A. M. et al. TSA: Micromesistius poutassou, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GHRY00000000.1 (2020).
Machado, A. M. et al. TSA: Trisopterus luscus, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GHRZ00000000.1 (2020).
Machado, A. M. et al. TSA: Scomber scombrus, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GHRT00000000.1 (2020).
Machado, A. M. et al. TSA: Trachurus trachurus, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GHRS00000000.1 (2020).
Machado, A. M. et al. Liver transcriptome resources of four commercially exploited teleost species. figshare https://doi.org/10.6084/m9.figshare.9033965.v3 (2020).
Mamrot, J. et al. De novo transcriptome assembly for the spiny mouse (Acomys cahirinus). Sci. Rep. 7, 8996 (2017).
Orsini, L. et al. Daphnia magna transcriptome by RNA-Seq across 12 environmental stressors. Sci. Data 3, 160030 (2016).
MacManes, M. D. The Oyster River Protocol: a multi-assembler and kmer approach for de novo transcriptome assembly. PeerJ 6, e5428 (2018).
Gioacchini, G. et al. A de novo transcriptome assembly approach elucidates the dynamics of ovarian maturation in the swordfish (Xiphias gladius). Sci. Rep. 9, 7375 (2019).
Maytin, A. K., Davies, S. W., Smith, G. E., Mullen, S. P. & Buston, P. M. De novo Transcriptome Assembly of the Clown Anemonefish (Amphiprion percula): A New Resource to Study the Evolution of Fish Color. Front. Mar. Sci. 5, 284 (2018).
Zhang, Q.-L. et al. Characterization of ladybird Henosepilachna vigintioctopunctata transcriptomes across various life stages. Sci. Data 5, 180093 (2018).
Onimaru, K., Tatsumi, K., Shibagaki, K. & Kuraku, S. A de novo transcriptome assembly of the zebra bullhead shark, Heterodontus zebra. Sci. Data 5, 180197 (2018).
Morandin, C., Pulliainen, U., Bos, N. & Schultner, E. De novo transcriptome assembly and its annotation for the black ant Formica fusca at the larval stage. Sci. Data 5, 180282 (2018).
Acknowledgements
This work was financed by the Project The Sea and the Shore, Architecture and Marine Biology: The Impact of Sea Life on the Built Environment [PTDC/ART-DAQ/29537/2017] from FCT/MCTES through National Funds (PIDDAC) and co-financing from the European Regional Development Fund (FEDER) [POCI-01-0145-FEDER-029537], in the aim of the new partnership agreement PT2020 through COMPETE 2020 – Competitiveness and Internationalization Operational Program (POCI), and by FCT – Fundação para a Ciência e a Tecnologia. This work is part of the CIIMAR-lead initiative Portugal-Fishomics. AM-M was supported by the project GenomePT (POCI-01-0145-FEDER-022184). R.R.F. thanks the Danish National Research Foundation (DNRF96) for its funding of the Center for Macroecology, Evolution, and Climate.
Author information
Authors and Affiliations
Contributions
L.F.C.C. supervised the study. A.M.M., L.F.C.C. and R.R.D.F. designed and conceived the work. M.F. collected the samples. E.F. performed the RNA extraction. A.M.M., A.M.-M., A.V. and R.P. conducted the transcriptome analyses. A.M.M., L.F.C.C., R.R.D.F., A.V. and E.F. wrote the first version of the manuscript. All authors revised and contribute to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Tables
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Machado, A.M., Muñoz-Merida, A., Fonseca, E. et al. Liver transcriptome resources of four commercially exploited teleost species. Sci Data 7, 214 (2020). https://doi.org/10.1038/s41597-020-0565-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0565-9
This article is cited by
-
The transcriptome assembly of the European freshwater mussel Unio elongatulus C. Pfeiffer, 1825
Scientific Data (2024)
-
The gill transcriptome of threatened European freshwater mussels
Scientific Data (2022)
