
Abstract
Pufferfish are ideal models for vertebrate chromosome evolution studies. The yellowbelly pufferfish, Takifugu flavidus, is an important marine fish species in the aquaculture industry and ecology of East Asia. The chromosome assembly of the species could facilitate the study of chromosome evolution and functional gene mapping. To this end, 44, 27 and 50 Gb reads were generated for genome assembly using Illumina, PacBio and Hi-C sequencing technologies, respectively. More than 13 Gb full-length transcripts were sequenced on the PacBio platform. A 366 Mb genome was obtained with the contig of 4.4 Mb and scaffold N50 length of 15.7 Mb. 266 contigs were reliably assembled into 22 chromosomes, representing 95.9% of the total genome. A total of 29,416 protein-coding genes were predicted and 28,071 genes were functionally annotated. More than 97.7% of the BUSCO genes were successfully detected in the genome. The genome resource in this work will be used for the conservation and population genetics of the yellowbelly pufferfish, as well as in vertebrate chromosome evolution studies.
Measurement(s) | whole genome sequencing assay • transcription profiling assay • sequence_assembly • sequence annotation |
Technology Type(s) | DNA sequencing • RNA sequencing assay • genome assembly • bioinformatics analysis |
Sample Characteristic - Organism | Takifugu flavidus |
Sample Characteristic - Environment | aquatic environment |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.10008740
Similar content being viewed by others


Background & Summary
The yellowbelly pufferfish (FishBase ID: 24266), Takifugu flavidus, is an economically and ecologically important fish species in coastal regions of East Asia, including the East China Sea, Yellow Sea and Bohai Bay1,2. The yellowbelly pufferfish is also a temperate bottom fish that exhibits only short-distance seasonal migration3. The yellowbelly pufferfish is caught and cultivated as a delicious fish species with high market value2,4,5. However, due to environmental deterioration and overfishing, the wild populations of the species have declined in the last decade6,7. Additionally, a low survival rate in artificial breeding has greatly limited the development of the marine aquaculture of the yellowbelly pufferfish8,9. Previous studies of the yellowbelly pufferfish have mainly focused on behavioural1, morphological and growth characteristics, temperature and salinity effects on embryos and larval development9, and molecular marker development10. A genome of Takifugu flavidus was published in 201411; however, this genome was a fragmented draft with contig and scaffold N50 lengths of 2.7 kb and 305.7 kb, respectively. A high-quality reference genome of the yellowbelly pufferfish could facilitate and prompt conservation genetics research and investigation of the molecular mechanisms of important economic traits of the species.
The genomes of pufferfish have also played an important role in studies of vertebrate genome evolution due to the compactness of genus Takifugu genomes12,13,14,15. Previous studies have shown that the number of repetitive elements in pufferfish is significantly reduced14,15. The genome of Takifugu rubripes, the other pufferfish in genus Takifugu, exhibits conserved linkages with humans, implying the preservation of chromosomal segments from the common vertebrate ancestor13,14,15. Another pufferfish genome, that of Tetraodon nigroviridis, was the second pufferfish genome reported14. Although the yellowbelly pufferfish genome has been published, the genome was constructed using short reads from SOLiD next-generation sequencing, and the sequences have not been assembled at the chromosomal level11. The elucidation of genomic evolution among pufferfish species, and comparison with other vertebrates such as humans, will require further chromosomal genome assemblies for pufferfish species.
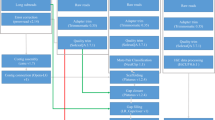
In this work, we applied a combined strategy involving Illumina, PacBio and Hi-C technologies to generate sequencing data for chromosomal genome construction for the yellowbelly pufferfish (Fig. 1). More than 98% of BUSCO genes were detected, and the contig and scaffold N50 lengths reached 4.4 and 15.7 Mb, respectively, demonstrating the outstanding completeness and sequence continuity of the genome. A total of 29,416 protein-coding genes were predicted in the assembled genome, and more than 95% of those genes were successfully functionally annotated. We believe that the chromosomal genome assembly constructed in this work will not only be valuable for ecology, conservation and aquaculture studies of the yellowbelly pufferfish but will also be of general interest in the evolutionary investigation of teleosts and vertebrates.

The work flow used for the yellowbelly pufferfish genome assembly and annotation in this work. The panes with green, cyan and yellow represent the input sequencing data, intermediate files and final outputs, respectively. Bioinformatics software is highlighted in red along the work flow.
Methods
Sample collection
A female yellowbelly pufferfish (Fig. 2), reared in the fish breeding centre of Fujian Normal University in Fuzhou City of Fujian Province was used for genome sequencing and assembly. Fresh white muscle, eye, skin, gonad, gut, liver, kidney, blood, gall bladder and air bladder tissues were collected and quickly frozen in liquid nitrogen for one hour. White muscle tissues were used for DNA sequencing for genome assembly, while all tissues were used for transcriptome sequencing.

A picture of the yellowbelly pufferfish used in the genome sequencing and assembly.
DNA and RNA sequencing
Genomic DNA from white muscle tissue was extracted using the standard phenol/chloroform extraction method for DNA sequencing library construction. The integrity of the genomic DNA molecules was checked using agarose gel electrophoresis. Both the Illumina HiSeq X Ten platform and the PacBio SEQUEL platform were applied for genomic sequencing to generate short and long genomic reads, respectively. For the Illumina X Ten platform (San Diego, CA, USA), a paired-end library was constructed with an insert size of 250 base pairs (bp) according to the protocol provided by the manufacturer. As a result, 44 Gb (~120X coverage of the estimated genome size, Table 1) of accurate short reads were generated, which were further cleaned using the HTQC utility16. Adapter sequences and reads with more than 10% N bases or low-quality bases (≤5) were filtered from the sequencing data. After filtering, 41.8 Gb (~110X, Table 1) of cleaned data were retained for the following analysis. To obtain sufficient sequencing data for genome assembly, we constructed two 20 kb DNA libraries using the extracted DNA and the standard Pacific Biosciences (PacBio, Menlo Park, CA) protocol, and fragments were selected using the Blue Pippin Size-Selection System (Sage Science, MA, USA). The library was sequenced using the PacBio SEQUEL platform. After removing adaptors, we obtained 27.2 Gb subreads (~73X, Table 1) for genome assembly. The genomic sequencing data used for subsequent genome assembly are summarized in Table 1.
We also performed RNA sequencing to generate transcriptome data for gene model prediction. To include as many tissue-specific transcripts as possible, multiple tissues were collected, as indicated in the Sample Collection section. TRIzol reagent (Invitrogen, USA) was used to separately extract RNA from all of the collected tissues, including white muscle, ocular, skin, gonad, intestine, liver, kidney, blood, gall bladder and air bladder tissues. RNA quality was checked with a NanoDrop ND-1000 spectrophotometer (Labtech, Ringmer, UK) and a 2100 Bioanalyzer (Agilent Technologies, CA, USA). Then, RNA molecules were equally mixed for transcriptome sequencing on the PacBio SEQUEL platform. First, cDNA was prepared using the SMARTer PCR cDNA Synthesis Kit (Clontech) from 1 μg of purified RNA. The Iso-Seq libraries were constructed from the BluePippin (Sage Science, MA, USA) size-selected cDNA with a size range of 0.6–3 kb according to the PacBio SEQUEL library construction protocol. Two SMRT flow cells were used for long-read transcriptome sequencing, and the resulting data used for gene prediction are summarized in Table 1.
De novo assembly of the yellowbelly pufferfish genome
For the Next Generation Sequencing (NGS) short reads, the Kmer-based method17 was used to perform genome survey analysis to estimate the genome size, heterozygosity and repeat content of the yellowbelly pufferfish genome. We counted the number of each 17-mer with Jellyfish18, and the frequency distribution is plotted in Fig. 3. The yellowbelly pufferfish genome size was then estimated from the frequency distribution to be 377 Mb.

The 17-mer count distribution for the genome size estimation. Note that the peaks around the depths of 33, 66 and 132 represent the heterozygous, homozygous and repeated Kmers, respectively.
The Falcon package19 was used to assemble the yellowbelly pufferfish genome with PacBio long reads, using the parameters length_cutoff = 10 kb and pr_length_cutoff = 8 kb. The procedures of long-read self-correction, corrected read alignment, sequence graph construction, and contig assembly were performed in Falcon. To correct random sequencing errors in the Falcon output, two steps of genome sequence polishing were applied: we first used arrow20 to polish the genome using long sequencing data, and two rounds of polishing using NGS short reads were then applied with Pilon21. Finally, we obtained a final contig assembly of 366 Mb with a contig N50 length of 4.4 Mb (Table 2).
To evaluate the quality of the assembled genome, the completeness and accuracy were assessed via BUSCO analysis and short-read mapping. The completeness of the assembled yellowbelly pufferfish genome was assessed by using BUSCO v3.022 with the vertebrata_odb9 database. We found that 95.7% and 2.7% of 2,586 BUSCO genes were completely and partially BUSCO genes were detected in the genome. We also aligned NGS short reads to the genome and found that more than 98.5% of the reads were reliably aligned, showing a high mapping ratio for the short-read sequencing data.
Chromosome construction using interaction information from Hi-C data
In this work, we applied the Hi-C technique for chromosome construction for the yellowbelly pufferfish. Although the Hi-C technique was first introduced to quantify genome-wide interactions23, the method exhibits suitability for chromosome assembly and has been successfully applied in many genomic projects24. In our study, we used 0.2 ml of blood from the same individual used for genome sequencing for Hi-C library construction and sequencing using the same method as in a previous study25,26. From the Hi-C library sequencing, approximately 50.7 Gb of data were generated (Table 1). The sequencing reads were mapped to the polished yellowbelly pufferfish genome with Bowtie 1.2.227. We independently aligned the two read ends to the genome and only selected the read pairs for which both ends were uniquely aligned to the genome. The hiclib Python library28 and a previously reported method24 were applied to filter the Hi-C reads, and the interaction frequency was quantified and normalized among contigs. Lachesis29 with default parameters was then applied to cluster contigs with the agglomerative hierarchical clustering method using the interaction matrix between sequences. Among the 169 million read pairs generated from Hi-C sequencing, 59 million read pairs (34.9%) provided valid interaction information for chromosome assembly. As a result, the contigs from the yellowbelly pufferfish were successfully clustered into 22 groups, which were further ordered and oriented into chromosomes. Finally, 271 contigs were reliably anchored on chromosomes, accounting for 95.9% of the total genome. The contig and scaffold N50 values reached 4.4 and 15.7 Mb (Tables 2 and 3), respectively, providing the first chromosomal genome assembly for the yellowbelly pufferfish.
Gene model prediction and functional annotations
Repeat elements were annotated in the yellowbelly pufferfish before gene model annotation. We applied Tandem Repeat Finder (TRF)30, LTR_FINDER31, PILER32 and RepeatScout33 for the ab initio prediction of repeat elements in the genome. Thereafter, RepeatMasker and RepeatProteinMask (http://www.repeatmasker.org) were used to search the genome sequences for known repeat elements, with the genome sequences used as queries against the Repbase database34. The repetitive element annotations are listed in Table 4.
Gene annotation was performed on the repetitive-element-masked genome. A combined strategy of homology-based, ab initio and transcriptome-based gene prediction methods was used. Protein sequences of Astyanax mexicanus, Danio rerio, Gadus morhua, Ictalurus punctatus, Oryzias latipes, Takifugu rubripes, Tetraodon nigroviridis and Oreochromis niloticus were downloaded from Ensembl35. Proteins from the closely related fish species were mapped to the yellowbelly pufferfish genome using TBLASTN36. The alignments were joined with Solar, and GeneWise37 was used to predict the exact gene structure of the corresponding genomic regions. Augustus38 was also used for the ab initio prediction of genes in the repeat-masked genome. Finally, the full-length transcriptome sequences generated from PacBio sequencing were aligned to the genome using the TopHat package39, and gene structure was predicted using Cufflinks40. All gene models were merged, and redundancy was removed by MAKER41, leading to a total of 29,416 protein-coding genes.
The NCBI non-redundant protein (nr) database and the SwissProt database with an E-value threshold of 1e-5 were used for the functional annotation of the protein-coding genes using BLASTX and the BLASTN utility42. Functional ontology and pathway information from the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases was assigned to the genes using Blast2GO43. Ultimately, 28,017 genes (95.24% of the total) of the yellowbelly pufferfish were functionally annotated (Table 5).
Data Records
The sequencing dataset and genome assembly were deposited in public repositories. The previous genome assembly can be accessed under accession number AOOT01000000 in NCBI44. The genomic Illumina sequencing data, genomic PacBio sequencing data, transcriptomic PacBio sequencing data, and the genomic Hi-C sequencing data were deposited as SRP162225 in NCBI45. The final chromosome assembly and genome annotation were submitted to NCBI Assembly with accession number RHFK0000000046. The functional annotation files are also available at figshare47.
Technical Validation
The quality of the DNA and RNA molecules and libraries used for genomic sequencing and transcriptome sequencing was validated before sequencing. The extracted DNA spectrophotometer ratios (SP) were 260/280 ≥1.6 for both Illumina and PacBio sequencing. DNA samples >2 μg and 20 μg were used for Illumina and PacBio sequencing, respectively. The concentration and quality of the total RNA were evaluated using a NanoVue Plus spectrophotometer (GE Healthcare, NJ, USA). RNA samples with a total RNA amount ≥10 μg, RNA integrity number ≥8, and rRNA ratio ≥1.5 were used to construct the sequencing library.
To validate our genome assembly, we compared the new genome to the previous genome. The new genome contained significantly fewer ambiguous bases (0.02 Mb) than the previous genome (67.9 Mb), but the size (366 Mb) of the new genome was approximately 20 Mb larger than that of the previous genome (346 Mb). Considering the estimated genome size of 377 Mb determined from the Kmer-based method, our new genome exhibited high completeness compared to the previous genome. The contig and scaffold N50 values of the newly assembled genome were almost 4,000 and 50 times higher than those of the previous genome, indicating a remarkable improvement in the sequence continuity of our assembly. We attributed the completeness and the continuity of the new genome to the application of PacBio long reads in the genome assembly. To further validate the improved continuity, we aligned genome fragments to our new genome with the NUCmer utility and found that more than 76% of the contigs were reliably mapped to the new genome with alignment ratios greater than 95%. Figure 4 provides two examples of alignments of genome sequences from the previous genome to our new assembly, showing that our new genome has significantly improved sequence continuity compared to the previous version.

Two examples of the alignment of scaffolds from the previous genome assembly to our new yellowbelly pufferfish genome assembly. (a) Alignments on contig5 in the new genome. (b) Alignment on contig74 in the new genome. The X axis represents the scaffolds from the previous genome, and the Y axis represents the contig sequences assembled in this work. The straight and reverse alignments of the scaffold sequences are shown in blue and red, respectively.
Usage Notes
The contig sequences were assembled into chromosomes using interaction information from Hi-C sequencing data; therefore we used 100 bp to represent the unknown gap sizes among contigs in the chromosome sequences.
Code availability
No specific code or script was used in this work. All commands used in the processing were executed according to the manual and protocols of the corresponding bioinformatics software.
References
Shi, Y. H. et al. Growth,development and behavior ecology of tawny puffer (Takifugu flavidus) larvae and juveniles. Journal of Fisheries of China 34, 1509–1517 (2010).
Zhong, J. X. et al. Studies on artificial propagation and larva-rearing of Fugu flavidus. Marine Sciences 33, 1–7 (2009).
Zhang, G., Shi, Y., Zhu, Y., Liu, J. & Zang, W. Effects of salinity on embryos and larvae of tawny puffer Takifugu flavidus. Aquaculture 302, 71–75, https://doi.org/10.1016/j.aquaculture.2010.02.005 (2010).
Jia-Bo, X. U. et al. Analysis of Lipid and Fatty Acid Composition in Different Tissues of Adult Female and Male Takifugu flavidus. Food Science 35, 133–137 (2014).
Tao, N. P., Wang, L. Y., Gong, X. & Liu, Y. Comparison of nutritional composition of farmed pufferfish muscles among Fugu obscurus, Fugu flavidus and Fugu rubripes. Journal of Food Composition & Analysis 28, 40–45 (2012).
Stump, E., Ralph, G. M., Comeros-Raynal, M. T., Matsuura, K. & Carpenter, K. Global conservation status of marine pufferfishes (Tetraodontiformes: Tetraodontidae). Global Ecology & Conservation 14, e00388 (2018).
Liu, Y., Qin, Z., Liu, H., Chao, L. & Tong, A. The complete mitochondrial genome sequence of Takifugu flavidus (Tetraodontiformes: Tetrodontidae). Mitochondrial Dna A Dna Mapp Seq Anal 27, 613–614 (2014).
Shi, Y. H., Zhang, G. Y., Zhu, Y. Z., Liu, J. Z. & Zang, W. L. Effects of temperature on fertilized eggs and larvae of tawny puffer Takifugu flavidus. Aquaculture Research 41, 1741–1747 (2010).
Shi, Y., Zhang, G., Liu, J. & Zang, W. Effects of temperature and salinity on oxygen consumption of tawny puffer Takifugu flavidus juvenile. Aquaculture Research 42, 301–307, https://doi.org/10.1111/j.1365-2109.2010.02638.x (2011).
Ma, H., Chen, S., Liao, X., Xu, T. & Ge, J. Isolation and characterization of polymorphic microsatellite loci from a dinucleotide-enriched genomic library of obscure puffer (Takifugu obscurus) and cross-species amplification. Conservation Genetics 10, 955–957, https://doi.org/10.1007/s10592-008-9540-2 (2009).
Gao, Y. et al. Draft Sequencing and Analysis of the Genome of Pufferfish Takifugu flavidus. DNA Research 21, 627–637, https://doi.org/10.1093/dnares/dsu025 (2014).
Gao, Y. et al. Draft Sequencing and Analysis of the Genome of Pufferfish Takifugu flavidus. DNA Research 21, 627–637 (2014).
Volff, J. N., Braasch, I. & Froschauer, A. Fish Genomes, Comparative Genomics and Vertebrate Evolution. Current Genomics 7, 43–57 (2006).
Jaillon, O. et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946 (2004).
Aparicio, S. et al. Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu rubripes. Science 297, 1301–1310 (2002).
Yang, X. et al. HTQC: a fast quality control toolkit for Illumina sequencing data. Bmc Bioinformatics 14, 1–4 (2013).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quantitative Biology 35, 62–67 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764 (2011).
Chin, C. S. et al. Phased Diploid Genome Assembly with Single Molecule Real-Time Sequencing. Nature Methods 13, 1050–1054 (2016).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563 (2013).
Walker, B. J. et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. Plos One 9, e112963 (2014).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210 (2015).
Belaghzal, H., Dekker, J. & Gibcus, J. H. HI-C 2.0: An optimized hi-c procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123, 56–65 (2017).
Nicolas, S. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, 259 (2015).
Xu, S. et al. A draft genome assembly of the Chinese sillago (Sillago sinica), the first reference genome for Sillaginidae fishes. GigaScience 7, giy108–giy108, https://doi.org/10.1093/gigascience/giy108 (2018).
Gong, G. et al. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. GigaScience 7, giy120–giy120, https://doi.org/10.1093/gigascience/giy120 (2018).
Langmead, B. Aligning short sequencing reads with Bowtie. (John Wiley & Sons, Inc., 2010).
Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods 9, 999 (2012).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology 31, 1119–1125 (2013).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573 (1999).
Zhao, X. & Hao, W. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351 (2005).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic & Genome Research 110, 462–467 (2005).
Flicek, P. et al. Ensembl 2014. Nucleic Acids Research 42, D749–D755 (2014).
Gertz, E. M. et al. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. Bmc Biology 4, 41 (2006).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Research 14, 988 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, 435–439 (2006).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Ghosh, S. & Chan, C. K. K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods in Molecular Biology 1374, 339 (2016).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research 18, 188–196 (2008).
Lobo, I. Basic Local Alignment Search Tool (BLAST). Journal of Molecular Biology 215, 403–410 (2008).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674 (2005).
Gao, Y. Takifugu flavidus, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:AOOT01000000 (2013).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP162225 (2018).
Xiao, S. Takifugu flavidus isolate HTHZ2018, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:RHFK00000000 (2018).
Xiao, S. Function annotation of Takifugu flavidus genome genes. figshare. https://doi.org/10.6084/m9.figshare.9944087.v1 (2019).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2018YFD0901102), the National Key Research and Development Program of China (2016YFC1200500) and the 13th Five-Year Plan for the Marine Innovation and Economic Development Demonstration Projects (FZHJ14). Z.H. was supported by the Special Fund for Marine Economic Development of Fujian Province (ZHHY-2019-3). S.X. was supported by the National Natural Science Foundation of China (Grant No. 31602207). Z.L. and J.Z. were supported by the Seed Industry Innovation and Industrialization Project of Takifugu (2017FJSCZY03). Y.C. was supported by a research grant from the Special Fund for Provincial Marine and Fishery Structural Adjustment from the Finance Department of Fujian Province (No. 1177[2017]).
Author information
Authors and Affiliations
Contributions
Z.H., X.H. and Y.C. conceived and designed the study; G.L., Z.L., J.Z., Y.Z. and Y.G. collected the samples; Y.Z., D.C. and T.X. performed DNA sequencing and Hi-C experiments; Y.X. and J.C. performed RNA sequencing; Y.Z., D.C. and S.X. estimated the genome size, assembled the genome, and assessed the assembly quality; S.X. and Y.Z. performed the genome annotation and functional genomic analysis. S.X., Z.H. and Y.Z. wrote the manuscript. All authors read, edited, and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Zhou, Y., Xiao, S., Lin, G. et al. Chromosome genome assembly and annotation of the yellowbelly pufferfish with PacBio and Hi-C sequencing data. Sci Data 6, 267 (2019). https://doi.org/10.1038/s41597-019-0279-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0279-z
This article is cited by
-
Improved high-quality reference genome of red drum facilitates the processes of resistance-related gene exploration
Scientific Data (2023)
-
High-quality wild barley genome assemblies and annotation with Nanopore long reads and Hi-C sequencing data
Scientific Data (2023)
-
Chromosome-level haplotype-resolved genome assembly for Takifugu ocellatus using PacBio and Hi-C technologies
Scientific Data (2023)
-
Chromosomal genome and population genetic analyses to reveal genetic architecture, breeding history and genes related to cadmium accumulation in Lentinula edodes
BMC Genomics (2022)
-
Telomere-to-telomere assembly of a fish Y chromosome reveals the origin of a young sex chromosome pair
Genome Biology (2021)
