
Abstract
Studies of naturally occurring cancers in dogs, which share many genetic and environmental factors with humans, provide valuable information as a comparative model for studying the mechanisms of human cancer pathogenesis. While individual and small-scale studies of canine cancers are underway, more generalized multi-omics studies have not been attempted due to the lack of large-scale and well-controlled genomic data. Here, we produced reliable whole-exome and whole-transcriptome sequencing data of 197 canine mammary cancers and their matched controls, annotated with rich clinical and biological features. Our dataset provides useful reference points for comparative analysis with human cancers and for developing novel diagnostic and therapeutic technologies for cancers in pet dogs.
Design Type(s) | disease state design • transcription profiling design • parallel group design |
Measurement Type(s) | transcription profiling assay • exome |
Technology Type(s) | RNA sequencing • DNA sequencing |
Factor Type(s) | breed • age • experimental condition • diagnosis |
Sample Characteristic(s) | Canis lupus familiaris • tissue |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
High-quality sequencing has clarified the dog genome with a coverage of >99%1,2. Moreover, the availability of a high-coverage reference genome and the emergence of higher-resolution next-generation sequencing have led to the identification of genomic structures coded by the dog genome, as provided in CanFam3.13. Based on such genomic information, cost-efficient next-generation sequencing has become available, allowing researchers to target specific coding regions and other regulatory elements for dogs. We suspect that whole-exome sequencing (WES) and whole-transcriptome sequencing (WTS) could be applied to discover single nucleotide variations (SNVs)4,5 and mutations causing diseases in dogs, such as in progressive retinal atrophy6.
Spontaneously occurring canine mammary gland tumors (CMTs) are of great interest to cancer researchers due to their clinical importance. CMTs are the most prevalent neoplasm in intact female dogs, and approximately 50% are malignant7,8. Interestingly, CMTs have been found to be promising cancer models with which to study human breast cancer due to their marked biological and clinical similarities9,10. Indeed, histopathological classification and histological grading of CMTs have been adopted from those of human breast cancer11,12,13 and, just as in humans, are actively used as prognostic indicators11,12,14. A recent multi-omics study of CMTs from 12 individual dogs characterized genomic features of two subtypes (simple carcinomas and complex carcinomas) and identified similarities and differences therein with human breast cancer15. To establish a better model and a more accurate profile of the molecular landscape of CMT, well-controlled multi-omics data for a larger cohort is desired.
Accordingly, to provide a useful resource for genomic analysis, we produced WES and WTS sequencing data from 197 and 158 dogs with CMTs, respectively. Among them, 185 of 197 of the WES (DNA-Seq) and 64 of the 158 WTS (RNA-Seq) specimens were matched with appropriate controls: buffy coats or normal mammary tissue for WES and normal mammary tissue for WTS were matched. Histopathological characteristics were evaluated in all tumor samples, including histopathological subtype, grade, and lymphatic invasion, and samples were annotated with corresponding sequencing data. In addition, immunohistochemical evaluation was performed in 189 samples to determine estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2) status. The raw sequencing data were aligned to the CanFam3.1 reference genome following the Best Practices announced by the Genome Analysis Toolkit (GATK, see Methods)16. We performed multiple quality control processes to confirm the quality of the sequencing and matched pairs of buffy coats and tumor tissues. Finally, the raw and aligned sequencing data, as well as normalized gene expression values (FPKM), were deposited in a public repository, along with the recorded clinical and biological information. A visual summary of the study design and workflow is shown in Fig. 1.

Overview of study design and workflow. (a) Study design of WES and RNA-Seq analysis. (b) Workflow of the WES data processing and variant calling. (c) Workflow of the RNA-Seq data processing. (d) Immunohistochemistry (IHC) workflow.
Overall, we are sharing a complete WES and WTS dataset that is ready for further biological analysis. We anticipate that this resource can be utilized for devising and validating various hypotheses in studies of comparative oncology between canine and human cancers.
Methods
Sample collection and preparation
Fresh and formalin-fixed paraffin-embedded (FFPE) tumor tissue samples of spontaneously occurring canine mammary tumors, adjacent normal tissue samples, and blood samples were obtained from privately-owned pet dogs via private veterinary clinics with informed consent the owners. Tissue samples were obtained as a part of routine diagnostic procedures, and blood samples were collected for research following the guidelines of and approval from the Institutional Animal Care and Use Committee of Konkuk University (KU16106 and KU17162). Fresh tissue samples were immediately transferred to RNAlater™ (Thermo Fisher Scientific, Vilnius, Lithuania), refrigerated overnight at 4 °C, and then stored at −80 °C until ready for analysis. For histopathology and immunohistochemistry (IHC) analysis, tissue samples were fixed in 10% neutral buffered formalin, processed routinely and embedded in paraffin wax. Blood samples were centrifuged, and buffy coats were isolated and stored at −80 °C until required for DNA extraction.
Genomic DNA was extracted from tissues using QIAamp DNA mini kits (Qiagen, Germany), and total RNA was extracted from tissues using RNeasy mini kits (Qiagen). Buffy coat DNA was extracted using QIAamp DNA blood mini kits (Qiagen) according to the manufacturer’s instructions.
Histopathology
Sections (4-μm thick) from the FFPE blocks were stained with hematoxylin and eosin and were diagnosed by veterinary pathologists (B.J.S. and J.H.S.). Histological subtype was determined by the World Health Organization classification11. Histological grade was assessed according to Peña system17, exclusively on the neoplastic epithelial component. In the case of mammary osteosarcoma and mammary fibrosarcoma, histological grade was assessed according to the grading system for canine osteosarcoma18 and the grading system for cutaneous and subcutaneous soft tissue sarcoma in dogs19, respectively. Lymphatic invasion, defined as infiltration of tumor cells in peritumoral lymphatic vessels (all cases) or infiltration of regional lymph nodes (only available cases), was also assessed.
Immunohistochemistry
Formalin-fixed paraffin-embedded canine mammary tumor samples (except osteosarcoma, fibrosarcoma, and poorly fixed tissues) underwent detection of estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2) by IHC with primary antibodies for ER (Biogenex, San Ramon, CA, USA) and HER2 (Dako, Glostrup, Denmark). Immunohistochemistry was performed as described in the previous publication20. Adjacent normal mammary gland or mammary hyperplasia were used as positive controls for ER antibody. Control slides known to be positive for HER2 were used as controls for HER2 antibody. Isotype-matched antibodies were used as negative controls.
ER and HER2 status were evaluated by the two veterinary pathologists mentioned above. Only epithelial tumor cells of representative areas were evaluated. Expression of ER was evaluated based on guidelines suggested by Pena et al.17. Expression of HER2 was measured based on recent guidelines recommended by the American Society of Clinical Oncology/College of American Pathologists21. Due to observation of non-specific cytoplasmic staining (according to human criteria) in canine tissues, as described by Burrai et al.22, only membrane stains were considered for scoring in this study.
Whole-exome sequencing
We sequenced 197 samples following the Illumina HiSeq 2500 protocol outsourced to Theragenetex. Two hundred nanograms of fragmented DNA was prepared to construct libraries with the SureSelect Canine All Exon Kit (Agilent, Inc., USA) using the manufacturer’s protocol. Briefly, qualified genomic DNA samples were randomly fragmented by Covaris, followed by adapter ligation, purification, hybridization, and PCR. Captured libraries were subjected to an Agilent 2100 Bioanalyzer to evaluate quality and were loaded on to the Illumina HiSeq sequencer, according to the manufacturer’s recommendations.
RNA sequencing
Before library construction, RNA 6000 Nano kits (Agilent Technologies, CA) were used to assess RNA quality. For cDNA library construction, 1 ug of RNA was obtained and purified with oligo-dT magnetic beads. Fragmentation was performed with purified mRNA, and double-stranded cDNAs were synthesized. The cDNAs were primed with poly-A, and sequencing adapters were connected using TruSeq RNA sample prep kits (Illumina, CA). Fragments were filtered to a specific length using BluePippin 2% agarose gel cassettes (Sage Science, MA), and PCR amplification was conducted. Fragment lengths and quality were electrophoretically verified with Agilent High Sensitivity DNA kits (Agilent Technologies, CA). Libraries were observed with a window spanning an average of 392 bp, standard deviation of 66. Finally, Illumina HiSeq 2500 was used for sequencing (Illumina, CA).
Processing of whole-exome sequencing data
Sequences were aligned to the CanFam3.1 reference genome with BWA-MEM223 and were output in a technology-independent SAM/BAM reference file format. Next, duplicate fragments were marked and eliminated with Picard (version 2.2) (http://picard.sourceforge.net). After assessing mapping quality and filtering out low-quality mapped reads, paired read information was evaluated to ensure that all mate-pair information was in sync between each read. Then, processes of removing PCR duplicates, indel realignment, fixing mate information, base quality score recalibration, and variant quality score recalibration on putative SNVs and indels were performed.
Germline and somatic mutations were called from the alignment files using GATK4.024 following GATK Best Practices recommendations, with using CanFam3.1 (Ensembl Release 91) as reference: the whole pipeline was implemented in-house (see Code Availability). The VCF file produced by the pipeline uses reference bases on the positive strand of CanFam3.1 in the REF field, and variants are shown in the ALT field. We calculated the depth of coverage using GATK and then followed the typical XHMM workflow for CNV calls25.
Processing of RNA sequencing data
RNA-Seq data of 158 tumor samples and 64 normal samples matched with tumors were sequenced. Prepared reads (FASTQ files) were mapped to the canine reference genome CanFam3.1 (Canis lupus familiaris) using TopHat (v.2.0.9), with Ensembl gene annotation and fr-firststrand library type. FPKM (Fragments Per Kilobase of Transcript per Million) values were calculated by Cufflinks (v2.1.1) using aligned bam files.
Data Records
The raw FASTQ files of the WES and RNA-Seq data produced by Illumina Highseq. 2500 are available from the Sequence Read Archive26,27. The RAW FASTQ files and FPKM values of RNA-Seq are available in the Gene Expression Omnibus database28. All steps used to process the raw files in order to create the final file are available at our GitHub repository (see Code availability). Sample characteristics are summarized in Online-only Table 1. Details on age, neuter status, histopathology descriptions, and immunohistochemical evaluation are deposited at Figshare29. Additional metadata links to SRA and GEO with clinical information are provide at Figshare29. The VCF files for germline mutations (SNPs and indels) of 197 CMTs and 185 normal samples called by GATK haplotype caller and for somatic variant calls of whole exome sequencing of 185 matched CMT and normal samples called by Mutect2 can be accessed at Figshare29.
Technical Validation
Quality validation
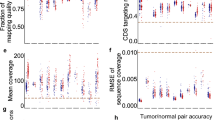
We validated quality of sequencing following the previous reported QC measures30. We used FASTQC v0.10.1 to analyze data quality via several measures, including sequence quality per base, GC content per sequence, sequence duplication levels, and quality score distribution over all sequences in the FASTQ files29. We randomly selected sample CMT-193 as a representative sample. Representative summary plots are provided in Fig. 2. High quality scores per base were shown, having a median quality score more than 30 both in WES (Fig. 2a left column) and RNA-Seq (Fig. 2b left column). The average quality score for overall sequences showed high scores above 30. Those score measures indicate that a large amount of the sequences in a run had high quality. The GC content of any strays were less than 5% in WES showing systemic bias free in sequence library (Fig. 2a middle column). The GC contents were uniform mostly although there were some bias in 1~8 bp in RNA-Seq (Fig. 2b). Examining sequence bias during polymerase chain reaction (PCR) amplification, we found that less than 2% of sequences were shown over 10 times in both platforms although RNA-Seq data have higher duplication rate than WES (Fig. 2a,b right column). We analyzed the quality score distribution of all sequences to verify if a subset of sequences had globally good quality. We applied Qualimap v2.2 to examine quality of sequencing alignment data according to features of the mapped reads. Qualimap highlights random errors and systematic biases, including PCR problems, GC content bias, and read contamination31. Mean mapping quality was around 60, and mean coverage was around 150X in WES (Fig. 3a). All other FASTQC and Qualimap files were shown to have quality metrics similar to those for randomly selected sample CMT-193. We calculated coverage using the “DepthOfCoverage” function in GATK and then calculated the percentage of bases with at least 100X and 200X coverage (Fig. 3b).

Confirmation of sequencing quality. (a) Quality score, GC content across all bases, and sequence duplication level in CMT-193 tumor (WES). (b) Quality score, GC content across all bases, and sequence duplication level in CMT-193 tumor (RNA-Seq).

Confirmation of sequencing quality and coverage. (a) Mapping quality and coverage in CMT-193 tumor (WES). 100X and 200X coverage in WES. Each bar on the x-axis represents a single sample, and the percentage on the y-axis indicates the percentage of bases out of all sequenced bases.
Concordance and swap for matched tumor–normal pairs
We checked concordance and swap to identify abnormal patterns of samples with large numbers of somatic mutations. We compared germline mutations in all samples pairwise with the following conditions: total allele depth >10, reference allele depth >=90% for genotypes (0/0), reference allele depth >=40%, reference allele depth <60% for genotypes (0/1), and alternative allele depth >=90% for genotypes (1/1). We calculated concordance ratios between all pair samples. Most alleles of tumor-normal pairs with the same sample ID were best matched; however, many abnormal normal samples had higher concordance with other unpaired tumor samples. We compared germline mutations in abnormal samples among the WES with RNA-Seq data and found high concordance between platforms for the same sample IDs. From the analysis, we found 23 unmatched pairs and the possibility of swapping for buffy coats. We excluded them from the downstream analysis. Among unmatched paired samples, we re-sequenced 11 normal samples whose normal tissues were available.
Sequence artifacts during shearing
Next-generation sequencing can produce sequence context-dependent artifacts, such as oxidation of guanine to 8-oxoguanine (OxoG) and FFPE deamination during genomic library preparation32. OxoG artifacts stem from oxidation of guanine to 8-oxoguanine, which results in G to T transversions. FFPE artifacts might be caused by formaldehyde deamination of cytosines, which results in C to T transitions. We ran the GATK4 “FilterByOrientationBias” function on Mutect2 calls and ensured that there was no OxoG or FFPE deamination in our dataset. Additionally, we manually checked whether samples had high C to A versus G to T conversion ratio.
Usage Notes
The bioinformatics pipeline used on our dataset, as outlined in Fig. 1, was mostly carried out using freely available and open access tools. Additionally, we conducted quality control analyses at multiple steps due to the possibilities of sample swapping and relatively poor standardization of canine analysis pipelines.
Detailed histology descriptions and IHC results of canine mammary tumors are described at Figshare29. Despite limitations in molecular classification in dogs and non-specific staining (HER2) in this study, our data will be helpful to further canine mammary tumor studies.
The size and the composition of the deposited dataset are subject to change according to further quality control and additional sequencing. The updated information will be noted in the corresponding data repository sites.
Code Availability
A full description of our analysis pipeline, describing all of the programs and parameters used, is openly available at https://github.com/irobii/cmt. The Markdown file in the pipeline folder documents each step of the pipeline, as well as provides external links to relevant sources for further information.
References
Lindblad-Toh, K. et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819 (2005).
Hoeppner, M. P. et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS One 9, e91172 (2014).
van Steenbeek, F. G., Hytonen, M. K., Leegwater, P. A. & Lohi, H. The canine era: the rise of a biomedical model. Anim Genet 47, 519–527 (2016).
Bai, B. et al. DoGSD: the dog and wolf genome SNP database. Nucleic Acids Res 43, D777–783 (2015).
Wang, Y. et al. GSA: Genome Sequence Archive<sup/>. Genomics Proteomics Bioinformatics 15, 14–18 (2017).
Ahonen, S. J., Arumilli, M. & Lohi, H. A CNGB1 frameshift mutation in Papillon and Phalene dogs with progressive retinal atrophy. PLoS One 8, e72122 (2013).
Moe, L. Population-based incidence of mammary tumours in some dog breeds. J Reprod Fertil Suppl 57, 439–443 (2001).
Sleeckx, N., de Rooster, H., Veldhuis Kroeze, E. J., Van Ginneken, C. & Van Brantegem, L. Canine mammary tumours, an overview. Reprod Domest Anim 46, 1112–1131 (2011).
Pinho, S. S., Carvalho, S., Cabral, J., Reis, C. A. & Gartner, F. Canine tumors: a spontaneous animal model of human carcinogenesis. Transl Res 159, 165–172 (2012).
Ranieri, G. et al. A model of study for human cancer: Spontaneous occurring tumors in dogs. Biological features and translation for new anticancer therapies. Crit Rev Oncol Hematol 88, 187–197 (2013).
Misdorp, W., Else, R. W., Hellmén, E. & Lipscomb, T. P. Histological Classification of Mammary Tumors of the Dog and the Cat, Vol. 7, 11–29 (Armed Forces Institute of Pathology in cooperation with the American Registry of Pathology and the World Health Organization Collaborating Center for Worldwide Reference on Comparative Oncology, 1999).
Pena, L., De Andres, P. J., Clemente, M., Cuesta, P. & Perez-Alenza, M. D. Prognostic value of histological grading in noninflammatory canine mammary carcinomas in a prospective study with two-year follow-up: relationship with clinical and histological characteristics. Vet Pathol 50, 94–105 (2013).
Elston, C. W. & Ellis, I. O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19, 403–410 (1991).
Goldschmidt, M., Pena, L., Rasotto, R. & Zappulli, V. Classification and grading of canine mammary tumors. Vet Pathol 48, 117–131 (2011).
Liu, D. et al. Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer Res 74, 5045–5056 (2014).
do Valle, I. F. et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinformatics 17, 341 (2016).
Pena, L. et al. Canine mammary tumors: a review and consensus of standard guidelines on epithelial and myoepithelial phenotype markers, HER2, and hormone receptor assessment using immunohistochemistry. Vet Pathol 51, 127–145 (2014).
Loukopoulos, P. & Robinson, W. F. Clinicopathological relevance of tumour grading in canine osteosarcoma. J Comp Pathol 136, 65–73 (2007).
Dennis, M. M. et al. Prognostic factors for cutaneous and subcutaneous soft tissue sarcomas in dogs. Vet Pathol 48, 73–84 (2011).
Seung, B. J. et al. CD204-Expressing Tumor-Associated Macrophages Are Associated With Malignant, High-Grade, and Hormone Receptor-Negative Canine Mammary Gland Tumors. Vet Pathol 55, 417–424 (2018).
Wolff, A. C. et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 31, 3997–4013 (2013).
Burrai, G. P. et al. Investigation of HER2 expression in canine mammary tumors by antibody-based, transcriptomic and mass spectrometry analysis: is the dog a suitable animal model for human breast cancer? Tumour Biol 36, 9083–9091 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Fromer, M. et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 91, 597–607 (2012).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP159481 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP159466 (2018).
Gene Expression Omnibus https://identifiers.org/geo:GSE119810 (2018).
Kim, K. K. et al. Whole-exome and whole-transcriptome sequencing of canine mammary gland tumors. figshare, https://doi.org/10.6084/m9.figshare.c.4543784.v1 (2019).
Seco-Cervera, M. et al. Small RNA-seq analysis of circulating miRNAs to identify phenotypic variability in Friedreich’s ataxia patients. Sci. Data 5, 180021 (2018).
Okonechnikov, K., Conesa, A. & Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 41, e67 (2013).
Acknowledgements
This research was supported by the Bio & Medical Technology Development Program (NRF-2016M3A9B6903439) through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT. KKKIM was additionally supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A2062110).
Author information
Authors and Affiliations
Contributions
H.P., H.N., J.H.S., J.C. and S.K. designed the study; B.J.S. and D.W.S. collected biological materials and clinical information; B.J.S. and J.H.S. assessed histological parameters and evaluated IHC results; K.K.K., D.K., D.L. and H.N. analyzed the genomic and transcriptome data; J.H.S. and H.P. reviewed experimental data; S.L. reviewed the process of initial data processing and analysis; K.K.K., B.J.S., D.K. and S.K. wrote the manuscript. All authors approved the final draft.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Kim, KK., Seung, BJ., Kim, D. et al. Whole-exome and whole-transcriptome sequencing of canine mammary gland tumors. Sci Data 6, 147 (2019). https://doi.org/10.1038/s41597-019-0149-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0149-8
This article is cited by
-
CanISO: a database of genomic and transcriptomic variations in domestic dog (Canis lupus familiaris)
BMC Genomics (2023)
-
Human basal-like breast cancer is represented by one of the two mammary tumor subtypes in dogs
Breast Cancer Research (2023)
-
1H NMR based urinary metabolites profiling dataset of canine mammary tumors
Scientific Data (2022)
-
Protein Expression of PI3K/AKT/mTOR Pathway Targets Validated by Gene Expression and its Correlation with Prognosis in Canine Mammary Cancer
Journal of Mammary Gland Biology and Neoplasia (2022)
-
Identification of Metastasis-Associated Genes in Cutaneous Squamous Cell Carcinoma Based on Bioinformatics Analysis and Experimental Validation
Advances in Therapy (2022)
