
Abstract
Population genetic data from nuclear DNA has yet to be synthesized to allow broad scale comparisons of intraspecific diversity versus species diversity. The MacroPopGen database collates and geo-references vertebrate population genetic data across the Americas from 1,308 nuclear microsatellite DNA studies, 897 species, and 9,090 genetically distinct populations where genetic differentiation (FST) was measured. Caribbean populations were particularly distinguished from North, Central, and South American populations, in having higher differentiation (FST = 0.12 vs. 0.07–0.09) and lower mean numbers of alleles (MNA = 4.11 vs. 4.84–5.54). While mammalian populations had lower MNA (4.86) than anadromous fish, reptiles, amphibians, freshwater fish, and birds (5.34–7.81), mean heterozygosity was largely similar across groups (0.57–0.63). Mean FST was consistently lowest in anadromous fishes (0.06) and birds (0.05) relative to all other groups (0.09–0.11). Significant differences in Family/Genera variance among continental regions or taxonomic groups were also observed. MacroPopGen can be used in many future applications including latitudinal analyses, spatial analyses (e.g. central-margin), taxonomic comparisons, regional assessments of anthropogenic impacts on biodiversity, and conservation of wild populations.
Design Type(s) | species comparison design • individual genetic characteristics comparison design • data integration objective |
Measurement Type(s) | genetic variation |
Technology Type(s) | digital curation |
Factor Type(s) | Taxon • Species |
Sample Characteristic(s) | population of organisms |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background and Summary
Collating large quantities of data is useful not only for assessing large-scale patterns but also for testing theories, informing conservation initiatives, and providing a valuable resource for future data comparisons. In particular, macro-ecological biodiversity assessments are becoming increasingly popular to identify hotspots of species biodiversity that can inform local management strategies1,2,3,4,5. However, populations, not species, are generally recognized as the appropriate scale for the management of sustainable harvesting and protection in endangered species legislation6,7,8. Nevertheless, population diversity – the number of genetically distinct populations within species – is typically excluded from most biodiversity syntheses and large-scale conservation planning (e.g.1,9,10,11,12,13). This has consequences when assessing biodiversity loss, as population extinction occurs at a much faster rate than species loss, and as such, a species’ vulnerability could be grossly misrepresented14.
Molecular markers provide an increasingly effective way to differentiate populations and estimate population diversity15. One example is the global population diversity estimate based on allozymes and restriction fragment length polymorphisms where authors found on average 220 populations per species and estimated annual loss of 16 million populations, a coarse estimate obtained by dividing the number of sampling locations by the sampling area10. The collated data from this study was not made publicly available for future usage and is outdated following the advancement of genetic tools. No study has formally revisited these concepts since this 199710 study (16,17,18, but see14,19,20 for exceptions), indicating the need for collating population information.
Population genetic technologies have seen advances in recent years, switching from allozymes to microsatellites to single nucleotide polymorphisms (SNPs), largely due to the better resolution of within-population variation that more recent technologies provide15,21. Population structure studies and vulnerability assessments have used microsatellites as their molecular marker for the past two decades, yet this wealth of data has not been thoroughly collated, although a few authors have collated related information in the form of microsatellite genetic variation9,12, population density estimates11, and pairwise FST estimates13. Despite the great degree of data collation across these studies, no work has combined the geo-referencing of population-specific genetic variation, FST measurements, and the number of populations within a species to create a single database across a wide variety of taxa and geographic regions.
Here we provide the first description of the release of the Macro-ecological, Population Genetics Database (MacroPopGen Database) – a database that contains geo-referenced population-specific characteristics based on nuclear DNA microsatellites. It contains information on 897 species from 1,308 studies published between 1994–2017, and 9,090 distinct populations of amphibians, birds, fish [anadromous, brackish, catadromous, or freshwater], mammals, and reptiles, totalling 561,605 genotyped individuals. Every population entry is georeferenced to permit large-scale spatial analyses, opening a variety of opportunities for overlaying microsatellite genetic data with environmental, geographic, or anthropogenic variables. It allows for population diversity and FST to be directly compared to species and genetic diversity (e.g. heterozygosity and mean number of alleles) through mapping applications.
MacroPopGen exemplifies the importance and usefulness of collating population genetic data by standardizing data from >1000 different studies, allowing for large-scale comparisons and many future applications, including latitudinal analyses, spatial or temporal analyses, taxonomic comparisons and regional assessments of genetic diversity across taxa or in relation to anthropogenic effects. Previous works focusing on older markers have already shown incredible usefulness in testing a variety of genetic and ecological theories1,2,9. We provide a baseline database for future works to build from and to compare to, particularly for comparing results to different, newer technologies. We urge future population studies using newer technologies to strive for a similar standardized repository for reporting population-specific statistics.
Methods
Data collection
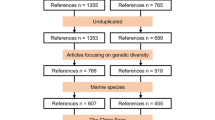
To collect population-genetic data from vertebrate populations located in the Americas, we first scanned Web of Science and Google Scholar for relevant articles using key search terms including country of occurrence, species common names, author names, and scientific names in combination with “microsatellite”, “distinct population”, and/or “FST”. A full list of the 1304 key terms and combinations used can be found online22. We also cross-referenced the list of bird microsatellite papers from Willoughby et al.9.
Search results with over 1000 hits would be filtered where if two consecutive pages did not yield a relevant result, further pages would not be considered (on average the first 15 pages on Google Scholar would be filtered for relevant articles). This preliminary screening limited results down to 6,297 peer-reviewed studies, technical reports, dissertations and government documents, of which only 1,308 fulfilled our criteria, including 142 of which were obtained from Willoughby et al.’s9 bird reference list. Once a study was selected, we extracted where possible: population locality name, latitude-longitude coordinates, average population-specific FST (Wright’s FST or Weir & Cockerham’s unbiased FST estimator θFST23,24), population-specific observed and expected heterozygosity averaged across loci (HO/HE, respectively), sample size (N), population-specific mean number of alleles per loci (MNA), study-specific corrected allelic richness (AR), and the number of microsatellite loci used in the study. For each population, we also documented the taxonomic group (amphibians, birds, fish [anadromous, brackish, catadromous, or freshwater], mammals, or reptiles), family, genus, species, common name, continent, and country. We chose not to include marine species because microsatellites have typically been unable to detect fine-scale population structure in such species, in contrast to the increased power and resolution of more recent genome-scale analyses for such species25. Instead we focus on terrestrial and aquatic ecosystems.
All populations were georeferenced in decimal degrees; if coordinates were not provided, they were inferred from the text or maps in a study. To calculate a metric of population-specific FST, we consulted pairwise FST tables and averaged across values that included the focal population, or population group if there was no significance between one or more population pairs. When only a global or regional FST was reported then that value would be used for all populations within the study; such FST values are indicated in the database where applicable.
Inclusion criteria and assumptions
A study was retained if two criteria were met: 1) microsatellites were used as molecular markers and 2) genetic differentiation was measured by Weir and Cockerham’s pairwise FST as opposed to other differentiation estimators because of its wide usage. Microsatellites were favoured over other molecular markers (e.g. SNPs, mitochondrial DNA, allozymes, RAPD, etc.) because their polymorphic nature allows them to resolve population structure at fine scales, particularly for closely related populations26,27. Additionally, microsatellites have higher mutation rates than other markers21,28 and have been one of the most widely used genetic markers in recent decades21. Therefore, microsatellites presently provide an abundance of collectable data across taxa relative to more recent molecular developments associated with single nucleotide polymorphisms (SNPs) or barcoding. While barcoding can assess phylogenetic signals across populations and species, microsatellites allow for the comparison of genetic characteristics between populations such as heterozygosity and allelic diversity, which has been noted to indicate levels of inbreeding or adaptive potential29,30,31,32.
Studies were assumed to have used selectively neutral nuclear microsatellite loci unless otherwise indicated because microsatellites are located within non-coding regions of the genome33 and have relatively fast mutation rates33,34. Microsatellite loci are often selected based on their polymorphism due to these faster mutation rates, causing concern that microsatellites may bias measures of genetic diversity compared to whole DNA sequencing-based measures34,35. Polymorphism bias has also been recognized in studies using other genetic markers such as SNPs21,34,36,37, and will continue to present challenges in genetic studies. An inherent assumption of this database is that ascertainment bias is similar across all studies and taxa, and therefore comparable. Additionally, previous work9 has concluded that the number of loci and primer type (whether cross-species or focal species) were not important in explaining variability in genetic diversity, an indication that ascertainment bias may not be very significant for large quantities of microsatellite data such as this database. Regardless, we tested ascertainment bias with a subset of the database, as described below.
Demarcating Populations
Populations were considered genetically distinct above a threshold FST value of 0.02. FST was used as the statistical measure of differentiation because of its standardized and common use in the literature for measuring genetic differentiation. The chosen threshold was based on a previous analytical review38, which indicated that genetic differentiation is not negligible if FST ≥ 0.05, but an FST value as low as 0.01 can also denote statistically significant differentiation38. While lower values of FST (0.02 to 0.01) are sufficient to show significant genetic differentiation, such values are more relevant for distinguishing specific taxonomic groups, such as marine fish populations which exhibit more gene flow38,39. Freshwater and terrestrial species tend to experience lower rates of gene flow than marine species and therefore an FST threshold above 0.01 is more appropriate13,39. To avoid accepting biologically insignificant population differentiation (type I error) or rejecting biologically significant differentiation (type II error) when demarcating populations, we considered the significance of FST values where available. We ensured that any pairwise comparisons >0.02 were statistically significant; we also checked significance when FST was <0.02 and significance implied two separate populations despite a lower FST. We also accounted for sample sizes with respect to significant FST. If sample size was five or less (occurring <0.1% of all cases in this study) and populations were found to be significantly different, the populations were instead grouped as one unless an adequate biological explanation was provided (n = 5). Likewise, if sample size was very large (e.g. >50) but FST was <0.02, consideration would be taken to determine if the populations were significantly different given the statistical support large sample sizes provide (usually given by p-values in the specific study, n = 63 cases where n ≥ 50 but FST ≤ 0.02). Additionally, if multiple studies were conducted in the same location for the same species, data from the most recent study or the one with the most microsatellite loci was used (n = 268 populations were duplicates and removed). When FST tables were unclear (e.g. many low FST values and no significance given), we considered results from population structure analyses (e.g. STRUCTURE, BAPS, etc.) to make informed decisions about population structure.
Geographic Breadth
We also report (i) how differentiated each population is in relation to all other populations it was compared to by calculating the average FST between a focal population and all other populations within that study, and (ii) the number of populations included in the calculation as well as the geographic distance or breadth that they span. For example, low FST values resulting from only a few sampling locations (e.g. 5) in a small geographic region (e.g. 10 km) may have a different interpretation than low FST values across many (e.g. >10) sampling locations in a broad geographic range (e.g. 10,000 km). To estimate the geographic breadth that sampled populations cover, we obtained coordinates for each population including locations that had been combined into one population. These data were put into a separate file that contains 10,921 sampling localities. Next, we used custom code22 utilizing the R package geosphere to calculate the maximum, minimum, and mean distances in metres between all populations of a study; distances are reported in metres in the database. We additionally note how many sampling localities make up each population in the database and how coordinates were obtained/estimated for populations that encompass multiple sampling localities.
Statistical Analysis
To calculate mean genetic diversity for taxonomic groups and continental regions we used generalized linear mixed models (GLMMs) that accounted for the random effect of study, species, genus, and family. Fixed effects included either the taxonomic group, or the continental region. Beta distributions were used to model HO and FST (R package glmmTMB v 0.2.2.0) because both these response variables and distributions are bounded between zero and one with no exact zeros or ones. Gamma distributions were used for MNA (R package lme4 v 1.1-18-1) as MNA follows a positively right skewed distribution characteristic of gamma distributions. We then used the R package and function emmeans (v 1.2.3) to calculate the mean values while accounting for model structure. For the models that used beta distributions, we used the function back.emmeans (R package RVAideMemoire v0.9-69-3) to back transform estimates.
To compare the degree of variation in each taxonomic or continental group, we calculated the coefficient of variation grouped at the species level for HO, MNA, and FST. Mixed models using the gamma distribution and random effects of reference, genus, and family were constructed. We then used model selection to see which between taxonomic group or continental region best explained differences between groups.
We assessed trends of ascertainment bias related to microsatellite loci development using a subset focusing on North American mammalian data (n = 1579 populations, 73 species)22. In addition to the number of microsatellite loci, we obtained from 230 mammalian studies the number of species used to develop those loci (ranged from 1 to 7), and whether the species were focal (n = 384), non-focal (n = 545), or mixed (n = 692), as well as information on the senior author’s country of affiliation. Using IUCN descriptions for each species, we also determined whether the species was harvested and to what extent (no n = 317, low n = 957, or high n = 347), the species’ IUCN status (Least Concern n = 1335, Near Threatened n = 45, Vulnerable n = 193, Endangered n = 41, Critically Endangered n = 7), whether the species was of conservation concern (no n = 561, low n = 211, or high n = 849), charismatic (no n = 495, low n = 189, or high n = 937), or of economic value (no n = 602, low n = 887, or high n = 132). Extent of harvesting was determined by the degree of harvesting described in IUCN’s “Use and Trade” category: none (“no”), subsistence or local harvesting (“low”), or substantial commercial harvesting (“high”). Conservation concern was specified to account for species that may have a lower IUCN rank (e.g. Least Concern, LC) but still have populations at risk or aspects of their habitat at risk (e.g. 563 LC species were still of conservation concern and therefore considered as “low”); this was largely described in IUCN’s “Threats” and “Conservation Action” categories. Charisma of species was somewhat subjective as it was determined by how generally well-known the species was, and whether the species may be considered a nuisance which would negatively affect their charisma score (e.g. the coyote is well known but can be considered a pest and as such its score was “low”). Economic value of a species was determined by the “Use and Trade” section, where if the species was commercially harvested it would be considered to have economic value (“high”); if the harvest has declined or is relatively low, a species’ economic value was considered as “low”.
We tested the fixed effects and interactions among these factors for ascertainment bias as well as the random effects of reference, species, genus, and family. We used GLMMs, using a beta distribution for HO (R package glmmTMB) and a gamma distribution for MNA (R package lme4). Following Zuur et al.40 guidelines for forwards and backwards model selection, we used the likelihood ratio test to find significant factors for the HO and MNA models, respectively.
Code and Data Availability
The data and R code used for the analyses are available from FigShare22.
Data Records
Data from the MacroPopGen database is hosted at Figshare22 and can be downloaded as one XLSX file. It consists of 9,098 rows (distinct populations), and 24 columns. The columns include taxonomic identifiers (family, genus, species, common name), population locality information, and study-specific data (sample size, population-specific FST, observed and expected heterozygosity, mean number of alleles, standardized allelic richness, latitude and longitude coordinates, reference ID, and year).
An additional XLSX file containing the corresponding references for each reference ID, and the list of key terms used in searches is also available on Figshare22. Most of the references were published in English, although a minority are in Spanish.
Technical Validation
Geographic and taxonomic bias
Between 1994 and 2017, most population microsatellite data came from species studied in North America (85.1%, Table 1). Fish species were the most represented taxonomic group, making up 44.8% of the database (Table 1). Salmonid species made up 55.9% of fish population data and represented 25.0% of data across all taxa.
When accounting for model structure, mean population genetic diversity differed significantly between some continental regions for HO and MNA (Fig. 1). Populations of South American species had the lowest HO while Caribbean populations showed significantly lower MNA (Table 1, Fig. 1). Despite some significant differences, the range of mean population genetic diversity metrics among continental regions was limited, between 0.57 and 0.61 for HO, and 4.11 and 5.5 for MNA (Fig. 1). Continental population differences in FST were stronger than for genetic diversity metrics, wherein Caribbean populations showed significantly higher population-specific FST, suggestive of less gene flow overall for these populations. This result follows general island-mainland expectations where island populations tend to be more isolated than mainland populations41,42.

Coefficient of variation and mean values for observed heterozygosity (HO), mean number of alleles (MNA), and population-specific FST calculated to account for GLMM structure. Error bars represent standard error. Significant differences between groups indicated by letter grouping where groups sharing the same letter(s) are not significantly different from one another. (a,b) Coefficient of variation calculated across (a) taxonomic groups (circles) and (b) between continental regions (squares). (c–e) Mean (c) FST, (d) Ho, and (e) MNA calculated across taxonomic groups. (f–h) Mean (f) FST, (g) Ho, and (h) MNA calculated between continental regions.
Among taxonomic groups, populations of anadromous fish had statistically higher mean genetic diversity (MNA = 7.8), and lower average FST values (0.06) aside from birds (mean FST = 0.05) (Fig. 1), consistent with previous work12,13. Mammalian populations also had lower mean MNA than all other groups (Fig. 1). However, there were no significant differences in mean HO between taxonomic groups (Fig. 1).
Variation among taxonomic and continental groups
There were significant differences in the coefficient of variation for HO among taxonomic groups but not continental regions, with bird species showing the least variation (Fig. 1). There were no significant differences in the coefficient of variation for species MNA across taxonomic groups or continental regions (Fig. 1). For FST, the only statistical difference was for the coefficient of variation to be larger in North American species relative to species in other regions, i.e. no taxonomic group differences in FST variance were found (Fig. 1). More variance among taxonomic distinctions was observed when considering within-family and within-genera variance in genetic metrics (Fig. 2). For example, the mean family HO ranged between 0.07–0.88, while MNA ranged from 1.40–24.97, and mean FST ranged from 0.0008–0.72; genera averages had a similar range for both metrics.

Microsatellite observed heterozygosity (HO), mean number of alleles (MNA), and population-specific FST averaged across each vertebrate group. Colours indicate the taxonomic group each family or genus belongs to: dark green = amphibians, purple = birds, blue = fish, orange = mammals, light green = reptiles. Error bars represent standard error. (a,c,e) Ho, MNA, and FST are averaged across vertebrate families (n = 195). (b,d,f) Ho, MNA, and FST are averaged across vertebrate genera (n = 480).
Bias with microsatellite loci
We assessed how genetic diversity and the number of microsatellite loci employed in empirical research has changed over time using linear models (Fig. 3). There has been a significant trend for increasing number of loci per year (R2 = 0.07, p <0.001) as well as a weak increase in genetic diversity with year (HO: R2 = 0. 0.009, p <0.001 and MNA: R2 = 0.001, p = 0.003). Additionally, we evaluated bias with respect to the number of microsatellite loci and the degree of genetic variation in HO and MNA using funnel plots (Fig. 4) and linear models. The plots appear to be largely symmetrical and show little bias with respect to number of loci, indicating the data capture a reasonable degree of genetic variation for the number of loci used. Note that we could not use a formal funnel plot test such as the Egger test because we do not have variance for HO and MNA for each study. However, the number of microsatellite loci was a significant predictor in linear models for both HO and MNA (p <0.001 for both), although adjusted R2 values were very small (0.002 and 0.03, respectively).

Observed heterozygosity, mean number of alleles, and number of microsatellite loci for populations of each taxonomic group sampled between the years 1994 to 2017. (a–c) All vertebrate groups together; (d–f) only amphibian species; (g–i) bird species; (j–l) all fish species; (m–o) mammalian species; (p–r) reptile species. Linear models are indicated for significant relationships.

Funnel plots for all populations; y axis for both plots is the number of microsatellite loci, and (a) x axis is observed heterozygosity (HO) or (b) mean number of alleles (MNA). Vertical line represents the mean value.
Ascertainment bias
After model selection testing for ascertainment bias with respect to loci type and origin, only the interaction between level of harvesting and conservation concern as well as the random effects of reference, family, and genus were significant for the HO model (Table 2). For the MNA model, the significant factors only included the interaction between conservation concern and charisma, as well as the random effects for reference and genus. None of the factors associated with microsatellite bias were retained in model selection (i.e. number of species used to derive loci, whether those species were focal, non-focal, or mixed). These results are consistent with previous assessments9 but indicate that microsatellite loci and loci origin do not significantly affect genetic diversity metrics when analyzed across diverse taxa.
References
Miraldo, A. et al. An Anthropocene map of genetic diversity. Science 353, 1532–1535 (2016).
Schluter, D. & Pennell, M. W. Speciation gradients and the distribution of biodiversity. Nature 546, 48–55 (2017).
Gaston, K. J. Global patterns in biodiversity. Nature 405, 220–227 (2000).
Brum, F. T. et al. Global priorities for conservation across multiple dimensions of mammalian diversity. Proc. Natl. Acad. Sci. 114, 7641–7646 (2017).
Abell, R. et al. Freshwater Ecoregions of the World: A New Map of Biogeographic Units for Freshwater Biodiversity Conservation. Bioscience 58, 403 (2008).
Stephenson, R. L. Stock complexity in fisheries management: a perspective of emerging issues related to population sub-units. Fish. Res. 43, 247–249 (1999).
Government of Canada. Species at Risk Act. (2002).
United States Fish & Wildlife Service. Endangered Species Act of 1973 As amended through the 108th Congress. Endangered Species Act Of 1973 (2003).
Willoughby, J. R. et al. The reduction of genetic diversity in threatened vertebrates and new recommendations regarding IUCN conservation rankings. Biol. Conserv. 191, 495–503 (2015).
Hughes, J. B., Daily, G. C. & Ehrlich, P. R. Population diversity: Its extent and extinction. Science 278, 689–692 (1997).
Santini, L. et al. Global drivers of population density in terrestrial vertebrates. Glob. Ecol. Biogeogr. 27, 968–979 (2018).
DeWoody, J. A. & Avise, J. C. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J. Fish Biol. 56, 461–473 (2000).
Medina, I., Cooke, G. M. & Ord, T. J. Walk, swim or fly? Locomotor mode predicts genetic differentiation in vertebrates. Ecol. Lett. 21, 638–645 (2018).
Ceballos, G., Ehrlich, P. R. & Dirzo, R. Biological annihilation via the ongoing sixth mass extinction signaled by vertebrate population losses and declines. Proc. Natl. Acad. Sci. 114, E6089–E6096 (2017).
Allendorf, F. W. Genetics and the conservation of natural populations: Allozymes to genomes. Mol. Ecol. 26, 420–430 (2017).
He, F. & Hubbell, S. P. Species-area relationships always overestimate extinction rates from habitat loss: Supplementary Information. Nature 473, 368–371 (2011).
Costello, M. J., May, R. M. & Stork, N. E. Can we name Earth’s species before they go extinct? Science 339, 413–416 (2013).
Rybicki, J. & Hanski, I. Species-area relationships and extinctions caused by habitat loss and fragmentation. Ecol. Lett. 16, 27–38 (2013).
Ceballos, G. Mammal Population Losses and the Extinction Crisis. Science 296, 904–907 (2002).
World Wildlife Fund. Living Planet Report Canada: A national look at wildlife loss. World Wildl. Fund (2017).
Schlötterer, C. The evolution of molecular markers — just a matter of fashion? Nat. Rev. Genet. 5, 63–69 (2004).
Lawrence, E. R. et al. MacroPopGen Database: Geo-referenced population-specific microsatellite data across the American continents. figshare https://doi.org/10.6084/m9.figshare.7207514.v1 (2018).
Weir, B. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Wright, S. The genetical structure of populations. Ann. Eugen. 15, 323–354 (1951).
Corander, J., Majander, K. K., Cheng, L. & Merilä, J. High degree of cryptic population differentiation in the baltic sea herring Clupea harengus. Mol. Ecol. 22(11), 2931–2940 (2013).
Jarne, P. & Lagoda, P. J. L. Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 11, 424–429 (1996).
Angers, B. & Bernatchez, L. Combined use of SMM and non-SMM methods to infer fine structure and evolutionary history of closely-related brook charr (Salvelinus fontinalis, Salmonidae) populations from microsatellites. Mol. Biol. Evol. 15, 143–159 (1998).
Selkoe, K. A. & Toonen, R. J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 9, 615–629 (2006).
Hansson, B. & Westerberg, L. On the correlation between heterozygosity and fitness in natural populations. Mol. Ecol. 11, 2467–2474 (2002).
Jump, A. S., Marchant, R. & Peñuelas, J. Environmental change and the option value of genetic diversity. Trends Plant Sci. 14, 51–58 (2009).
Reed, D. H. & Frankham, R. Correlation between Fitness and Genetic Diversity. Conserv. Biol. 17, 230–237 (2003).
Fraser, D. J. et al. Population correlates of rapid captive-induced maladaptation in a stream fish. Evol. Appl. 12, https://doi.org/10.1111/eva.12649 (2019).
Wiehe, T. The effect of selective sweeps on the variance of the allele distribution of a linked multiallele locus: hitchhiking of microsatellites. Theor. Popul. Biol. 53, 272–283 (1998).
Väli, Ü., Einarsson, A., Waits, L. & Ellegren, H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol. Ecol 17, 3808–3817 (2008).
Ellegren, H. et al. Microsatellite evolution–a reciprocal study of repeat lengths at homologous loci in cattle and sheep. Mol. Biol. Evol. 14, 854–860 (1997).
Nielsen, R. Population genetic analysis of ascertained SNP data. Hum. Genomics 1, 218–224 (2004).
Clark, A. G., Hubisz, M. J., Bustamante, C. D., Williamson, S. H. & Nielsen, R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 15, 1496–1502 (2005).
Waples, R. S. & Gaggiotti, O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol 15, 1419–1439 (2006).
Waples, R. S. Separating the wheat from the chaff: Patterns of genetic differentiation in high gene flow species. J. Hered. 89, 438–450 (1998).
Zuur, A. F., Ieno, E. N., Walker, N. J., Saveliev, A. A. & Smith, G. M. Mixed Effects Models and Extensions in Ecology with R. Statistics for Biology and Health. https://doi.org/10.1007/978-0-387-87458-6 (Spring Science and Business Media, 2009).
Jaenike, J. R. A Steady State Model of Genetic Polymorphism on Islands. Am. Nat. 107, 793–795 (1973).
Frankham, R. Do island populations have less genetic variation than mainland populations? Heredity 78, 311–327 (1997).
Acknowledgements
Much work went into building this database and we would like to thank J.D. Lawrence, K. Levasseur-Bhatia, A. de Jaham, D. MacRae, and C. Clegg, for additional help in building the database, finding population coordinates, and helping collate references; M. Yates for statistical support. Additionally, we would like to thank two anonymous reviewers who gave constructive feedback on the manuscript. We would sincerely like to thank Dr. K.J. Monsen, Dr. T. Beacham, and Dr. J. Willoughby for sharing datasets with us that enabled completion of the database. This work was funded by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant, and a Quebec Centre for Biodiversity Science Seed Grant.
Author information
Authors and Affiliations
Contributions
E.R.L. initiated the database construction, collected data on North American fish, mammals, amphibians, reptiles, and South American mammals; she also combined all files from other contributors, checked data for consistency, ran statistical analyses, built the reference file, and wrote the paper. J.N.B. collected population data on North American fish, and South American fish, mammals, amphibians, and reptiles, assisted with collating reference information, and provided feedback on manuscript revisions. J.-M.M. completed statistical modeling, focusing on analyses for beta distributions and coefficients of variation; he also provided feedback on manuscript revisions. K.M. collected population data on North American mammals, fish, reptiles, and amphibians, and provided feedback on manuscript revisions. Z.W. collected population data on North American fish and provided feedback on manuscript revisions. T.B. and N.K. collected population data on bird species within the American continents and provided feedback on manuscript revisions. A.H. collected population data on North American mammal species and provided feedback on manuscript revisions. G.N. collected FST values for bird species and assisted with collating reference information. R.A.K. collected FST values for bird species and provided feedback on manuscript revisions. D.J.F. conceived the general idea of the study, collected population data on North American fish, and provided feedback on manuscript revisions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ISA-Tab metadata file
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Lawrence, E.R., Benavente, J.N., Matte, JM. et al. Geo-referenced population-specific microsatellite data across American continents, the MacroPopGen Database. Sci Data 6, 14 (2019). https://doi.org/10.1038/s41597-019-0024-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0024-7
This article is cited by
-
A dataset of genetic diversity studies in the China Seas
Scientific Data (2024)
-
CaliPopGen: A genetic and life history database for the fauna and flora of California
Scientific Data (2022)
-
Opportunities and challenges of macrogenetic studies
Nature Reviews Genetics (2021)
-
Authors’ Reply to Letter to the Editor: Continued improvement to genetic diversity indicator for CBD
Conservation Genetics (2021)
-
Expediency of Tetra- and Pentanucleotide Repeat Autosomal STR Markers for DNA Typing in Central Indian Population
Proceedings of the National Academy of Sciences, India Section B: Biological Sciences (2020)
