
Abstract
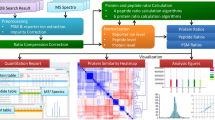
Cardinal v.3 is an open-source software for reproducible analysis of mass spectrometry imaging experiments. A major update from its previous versions, Cardinal v.3 supports most mass spectrometry imaging workflows. Its analytical capabilities include advanced data processing such as mass recalibration, advanced statistical analyses such as single-ion segmentation and rough annotation-based classification, and memory-efficient analyses of large-scale multitissue experiments.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


Data availability
The three publicly available datasets that were reanalyzed in this study are available via the PRIDE repository (https://www.ebi.ac.uk/pride/) with the identifiers PXD001283, PXD011104 and PXD026459. The raw, reanalysis and R code are available in the MassIVE.quant repository with the identifier MSV000086099 and RMSV000000684 (case study 1), MSV000086102 and RMSV000000664 (case study 2) and MSV000089594 and RMSV000000686 (case studies 3 and 4). Reanalysis and R code are also available in GitHub (https://github.com/Vitek-Lab/Cardinal3-vignettes).
Code availability
The following R (v.4.2.1 and 4.2.2) packages were used in this study: Cardinal v.3.1.0, CardinalNN v.0.0.1, reticulate v.1.28 and ggplot2 v.3.4.1. The following python (v.3.10.8) libraries were used in this study: classification (installed from DeepMSI) v.0.0.1, matplotlib v.3.6.2, numpy v.1.23.5, pandas v.1.5.2 and torch v.1.12.1. Code for the reanalysis of public datasets including generation of the figures is available on GitHub: https://github.com/Vitek-Lab/Cardinal3-vignettes. The Cardinal software can be downloaded via: Bioconductor, https://www.bioconductor.org/packages/release/bioc/html/Cardinal.html; Bioconda, https://anaconda.org/bioconda/bioconductor-cardinal; Biocontainers, https://biocontainers.pro/tools/bioconductor-cardinal; GitHub (Source code), https://github.com/kuwisdelu/Cardinal (Artistic 2.0) and https://github.com/DanGuo1223/CardinalNN (MIT license).
References
Spengler, B. Mass spectrometry imaging of biomolecular information. Anal. Chem. 87, 64–82 (2015).
Weiskirchen, R., Weiskirchen, S., Kim, P. & Winkler, R. Software solutions for evaluation and visualization of laser ablation inductively coupled plasma mass spectrometry imaging (LA-ICP-MSI) data: a short overview. J. Cheminform. 11, 16 (2019).
Klinkert, I., Chughtai, K., Ellis, S. R. & Heeren, R. M. A. Methods for full resolution data exploration and visualization for large 2D and 3D mass spectrometry imaging datasets. Int. J. Mass Spectrom. 362, 40–47 (2014).
Källback, P., Shariatgorji, M., Nilsson, A. & Andrén, P. E. Novel mass spectrometry imaging software assisting labeled normalization and quantitation of drugs and neuropeptides directly in tissue sections. J. Proteom. 75, 4941–4951 (2012).
Robichaud, G., Garrard, K. P., Barry, J. A. & Muddiman, D. C. MSiReader: an open-source interface to view and analyze high resolving power MS imaging files on Matlab platform. J. Am. Soc. Mass Spectrom. 24, 718–721 (2013).
Race, A. M. et al. SpectralAnalysis: software for the masses. Anal. Chem. 88, 9451–9458 (2016).
Ràfols, P. et al. RMSI: an R package for MS imaging data handling and visualization. Bioinformatics 33, 2427–2428 (2017).
Veselkov, K. et al. BASIS: high-performance bioinformatics platform for processing of large-scale mass spectrometry imaging data in chemically augmented histology. Sci. Rep. 8, 4053 (2018).
Cordes, J. et al. M2aia—interactive, fast, and memory-efficient analysis of 2D and 3D multi-modal mass spectrometry imaging data. GigaScience 10, giab049 (2021).
Guo, G. et al. Automated annotation and visualisation of high-resolution spatial proteomic mass spectrometry imaging data using HIT-MAP. Nat. Commun. 12, 3241 (2021).
Bond, N. J., Koulman, A., Griffin, J. L. & Hall, Z. massPix: an R package for annotation and interpretation of mass spectrometry imaging data for lipidomics. Metabolomics 13, 128 (2017).
Luu, G. T., Condren, A. R., Kahl, L. J., Dietrich, L. E. P. & Sanchez, L. M. Evaluation of data analysis platforms and compatibility with MALDI-TOF imaging mass spectrometry data sets. J. Am. Soc. Mass Spectrom. 31, 1313–1320 (2020).
Bemis, K. D. et al. Cardinal: an R package for statistical analysis of mass spectrometry-based imaging experiments. Bioinformatics 31, 2418–2420 (2015).
Bemis, K. A. et al. Statistical detection of differentially abundant ions in mass spectrometry-based imaging experiments with complex designs. Int. J. Mass Spectrom. 437, 49–57 (2019).
Guo, D., Bemis, K., Rawlins, C., Agar, J. & Vitek, O. Unsupervised segmentation of mass spectrometric ion images characterizes morphology of tissues. Bioinformatics 35, i208–i217 (2019).
Guo, D. et al. Deep multiple instance learning classifies subtissue locations in mass spectrometry images from tissue-level annotations. Bioinformatics 36, i300–i308 (2020).
Vizcaíno, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016).
Römpp, A. et al. Histology by mass spectrometry: label-free tissue characterization obtained from high-accuracy bioanalytical imaging. Angew. Chem. Int. Ed. 49, 3834–3838 (2010).
Erich, K. et al. Spatial distribution of endogenous tissue protease activity in gastric carcinoma mapped by MALDI mass spectrometry imaging. Mol. Cell. Proteomics 49, 151–161 (2018).
Föll, M. C. et al. Moving translational mass spectrometry imaging towards transparent and reproducible data analyses: a case study of an urothelial cancer cohort analyzed in the Galaxy framework. Clin. Proteom. 19, 8 (2022).
Choi, M. et al. MassIVE.quant: a community resource of quantitative mass spectrometry–based proteomics datasets. Nat. Methods 17, 981–984 (2020).
Bemis, K. D. et al. Probabilistic segmentation of mass spectrometry (MS) images helps select important ions and characterize confidence in the resulting segments. Mol. Cell. Proteom. 15, 1761–1772 (2016).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Grüning, B. et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476 (2018).
da Veiga Leprevost, F. et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics 33, 2580–2582 (2017).
Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351 (2022).
Föll, M. C. et al. Accessible and reproducible mass spectrometry imaging data analysis in Galaxy. GigaScience 8, giz143 (2019).
Patterson, N. H., Yang, E., Kranjec, E.-A. & Chaurand, P. Co-registration and analysis of multiple imaging mass spectrometry datasets targeting different analytes. Bioinformatics 35, 1261–1262 (2019).
Acknowledgements
We thank A. Weber for valuable feedback. This work was supported by NSF-BIO/DBI 1759736, NSF-BIO/DBI 1950412, NIH-NLM-R01 1R01LM013115 and the Chan-Zuckerberg foundation awarded to O.V., and the Hans A. Krebs Medical Scientist Programme, Faculty of Medicine, University of Freiburg, Germany awarded to M.C.F. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
K.A.B. developed the methods and wrote the code for the Cardinal and Matter software. D.G. developed the methods and wrote the code for the CardinalNN software. M.C.F. and S.S.L. tested all software. K.A.B., M.C.F. and D.G. developed and analyzed the data for the case studies and generated the vignettes. S.S.L. revised the vignettes. M.C.F. wrote the manuscript and generated the figures. M.C.F. and O.V. revised and edited the manuscript. O.V. supervised the work and provided funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Nathan Patterson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Arunima Singh, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Cardinal v.3 supports visualization and preprocessing of high mass resolution data.
a, Reproducing the overlay ion image of three m/z features with different spatial abundances (Fig. 1a in original publication). b, Zoomed in average mass spectra show two peaks that are only 0.05 m/z apart (Fig. 2c in original publication) and could be detected as separate peaks. The red dotted lines indicate that both peaks will be picked separately during peak detection. c, Visualizing the spatial distribution of both peaks shows that they are present in different tissue regions (Fig. 2b in original publication). The peaks in the original publication were likely mislabeled as suggested by Fig. S5 of the same publication and the inverse image we obtained here.
Extended Data Fig. 2 Cardinal v.3 enables more accurate m/z values in the multiple replicates classification dataset.
a, Zoomed in mass spectra for six random spectra of the first dataset show m/z shifts between the spectra before m/z alignment. Applying Cardinal’s new mass alignment step increases the alignment of the peaks substantially. b, Zoomed in mass spectra for six random spectra of the first dataset before and after mass re-calibration show how Cardinal’s new mass re-calibration method shifts the monoisotopic angiotensin peak towards its theoretical m/z position (m/z 1296.7, dashed vertical line).
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bemis, K.A., Föll, M.C., Guo, D. et al. Cardinal v.3: a versatile open-source software for mass spectrometry imaging analysis. Nat Methods 20, 1883–1886 (2023). https://doi.org/10.1038/s41592-023-02070-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-023-02070-z
