
Abstract
Host–microbe interactions have been linked to health and disease states through the use of microbial taxonomic profiling, mostly via 16S ribosomal RNA gene sequencing. However, many mechanistic insights remain elusive, in part because studying the genomes of microbes associated with mammalian tissue is difficult due to the high ratio of host to microbial DNA in such samples. Here we describe a microbial-enrichment method (MEM), which we demonstrate on a wide range of sample types, including saliva, stool, intestinal scrapings, and intestinal mucosal biopsies. MEM enabled high-throughput characterization of microbial metagenomes from human intestinal biopsies by reducing host DNA more than 1,000-fold with minimal microbial community changes (roughly 90% of taxa had no significant differences between MEM-treated and untreated control groups). Shotgun sequencing of MEM-treated human intestinal biopsies enabled characterization of both high- and low-abundance microbial taxa, pathways and genes longitudinally along the gastrointestinal tract. We report the construction of metagenome-assembled genomes directly from human intestinal biopsies for bacteria and archaea at relative abundances as low as 1%. Analysis of metagenome-assembled genomes reveals distinct subpopulation structures between the small and large intestine for some taxa. MEM opens a path for the microbiome field to acquire deeper insights into host–microbe interactions by enabling in-depth characterization of host-tissue-associated microbial communities.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
The datasets generated and analyzed during the current study are available at CaltechDATA, https://doi.org/10.22002/gx69z-wec80. Microbial sequencing data are available at NCBI Accession no. PRJNA991155. Sequencing data from human samples have been host scrubbed using STAT78 sra-human-scrubber (https://github.com/ncbi/sra-human-scrubber) followed by alignment to CHM13 (ref. 79). Source data are provided with this paper.
Code availability
The code used in data processing and analysis is available at CaltechDATA, https://doi.org/10.22002/gx69z-wec80.
References
Tuganbaev, T. et al. Diet diurnally regulates small intestinal microbiome-epithelial-immune homeostasis and enteritis. Cell 182, 1441–1459 (2020).
Dejea, C. M. et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597 (2018).
Bullman, S. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017).
Morgan, X. C. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79 (2012).
Caruso, R., Lo, B. C. & Nunez, G. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 20, 411–426 (2020).
Pascal, V. et al. A microbial signature for Crohn’s disease. Gut 66, 813–822 (2017).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014).
Cheng, J. et al. Duodenal microbiota composition and mucosal homeostasis in pediatric celiac disease. BMC Gastroenterol. 13, 113 (2013).
Earley, Z. M. et al. GATA4 controls regionalization of tissue immunity and commensal-driven immunopathology. Immunity 56, 43–57 (2023).
Ringel, Y. et al. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes 6, 173–181 (2015).
Parthasarathy, G. et al. Relationship between microbiota of the colonic mucosa vs feces and symptoms, colonic transit, and methane production in female patients with chronic constipation. Gastroenterology 150, 367–379 (2016).
Vaga, S. et al. Compositional and functional differences of the mucosal microbiota along the intestine of healthy individuals. Sci. Rep. 10, 14977 (2020).
Shen, T. D. et al. The mucosally-adherent rectal microbiota contains features unique to alcohol-related cirrhosis. Gut Microbes 13, 1987781 (2021).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2013).
Chen, L. X., Anantharaman, K., Shaiber, A., Eren, A. M. & Banfield, J. F. Accurate and complete genomes from metagenomes. Genome Res. 30, 315–333 (2020).
Vineis, J. H. et al. Patient-specific Bacteroides genome variants in pouchitis. mBio. 7, 10–1128 (2016).
Groussin, M., Mazel, F. & Alm, E. J. Co-evolution and co-speciation of host-gut bacteria systems. Cell Host Microbe 28, 12–22 (2020).
Wang, G. H., Dittmer, J., Douglas, B., Huang, L. & Brucker, R. M. Coadaptation between host genome and microbiome under long-term xenobiotic-induced selection. Sci. Adv. 7, eabd4473 (2021).
Tyson, G. W. et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37–43 (2004).
Pereira-Marques, J. et al. Impact of host DNA and sequencing depth on the taxonomic resolution of whole metagenome sequencing for microbiome analysis. Front. Microbiol. 10, 1277 (2019).
Marotz, C. A. et al. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6, 42 (2018).
Bruggeling, C. E. et al. Optimized bacterial DNA isolation method for microbiome analysis of human tissues. Microbiology Open 10, e1191 (2021).
Ganda, E. et al. DNA extraction and host depletion methods significantly impact and potentially bias bacterial detection in a biological fluid. mSystems 6, e0061921 (2021).
Avanzi, C. et al. Red squirrels in the British Isles are infected with leprosy bacilli. Science 354, 744–747 (2016).
Charalampous, T. et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 37, 783–792 (2019).
Cheng, W. Y. et al. High sensitivity of shotgun metagenomic sequencing in colon tissue biopsy by host DNA depletion. Genomics Proteomics Bioinformatics https://doi.org/10.1016/j.gpb.2022.09.003 (2022).
Oechslin, C. P. et al. Limited correlation of shotgun metagenomics following host depletion and routine diagnostics for viruses and bacteria in low concentrated surrogate and clinical samples. Front. Cell Infect. Microbiol. 8, 375 (2018).
Hasan, M. R. et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J. Clin. Microbiol. 54, 919–927 (2016).
Heravi, F. S., Zakrzewski, M., Vickery, K. & Hu, H. Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples. J. Microbiol. Meth. 170, 105856 (2020).
Shaffer, J. P. et al. A comparison of six DNA extraction protocols for 16S, ITS and shotgun metagenomic sequencing of microbial communities. BioTechniques 73, 34–46 (2022).
Hallmaier-Wacker, L. K., Lueert, S., Roos, C. & Knauf, S. The impact of storage buffer, DNA extraction method, and polymerase on microbial analysis. Sci. Rep. 8, 6292 (2018).
Bellali, S., Lagier, J. C., Raoult, D. & Bou Khalil, J. Among live and dead bacteria, the optimization of sample collection and processing remains essential in recovering gut microbiota components. Front. Microbiol. 10, 1606 (2019).
Barlow, J. T., Bogatyrev, S. R. & Ismagilov, R. F. A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities. Nat. Commun. 11, 2590 (2020).
Weyrich, L. S. et al. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 19, 982–996 (2019).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Velásquez-Mejía, E. P., de la Cuesta-Zuluaga, J. & Escobar, J. S. Impact of DNA extraction, sample dilution, and reagent contamination on 16S rRNA gene sequencing of human feces. Appl. Microbiol. Biotechnol. 102, 403–411 (2018).
Liu, Y., Elworth, R. A. L., Jochum, M. D., Aagaard, K. M. & Treangen, T. J. De novo identification of microbial contaminants in low microbial biomass microbiomes with Squeegee. Nat. Commun. 13, 6799 (2022).
Mehta, R. S. et al. Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 3, 347–355 (2018).
Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the Genome Taxonomy Database. Bioinformatics 38, 5315–5316 (2022).
Delmont, T. O. & Eren, A. M. Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 6, e4320 (2018).
Castrillo, G. et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 543, 513–518 (2017).
Fitzpatrick, C. R. et al. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl Acad. Sci. USA 115, E1157–E1165 (2018).
Shin, S. C. et al. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670–674 (2011).
Motta, E. V. S., Raymann, K. & Moran, N. A. Glyphosate perturbs the gut microbiota of honey bees. Proc. Natl Acad. Sci. USA 115, 10305–10310 (2018).
Poore, G. D. et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574 (2020).
Riquelme, E. et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795–806 (2019).
Olaisen, M. et al. Bacterial mucosa-associated microbiome in inflamed and proximal noninflamed ileum of patients with Crohn’s disease. Inflamm. Bowel Dis. 27, 12–24 (2021).
Liou, M. J. et al. Host cells subdivide nutrient niches into discrete biogeographical microhabitats for gut microbes. Cell Host Microbe 30, 836–847 (2022).
Libertucci, J. et al. Inflammation-related differences in mucosa-associated microbiota and intestinal barrier function in colonic Crohn’s disease. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G420–G431 (2018).
Brenchley, J. M. & Douek, D. C. Microbial translocation across the GI tract. Annu. Rev. Immunol. 30, 149–173 (2012).
Singer, J. R. et al. Preventing dysbiosis of the neonatal mouse intestinal microbiome protects against late-onset sepsis. Nat. Med 25, 1772–1782 (2019).
Maynard, C. L., Elson, C. O., Hatton, R. D. & Weaver, C. T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 489, 231–241 (2012).
Girdhar, K. et al. A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. Proc. Natl Acad. Sci. USA 119, e2120028119 (2022).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Ferreira, R. M. et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 67, 226–236 (2018).
Wilson, M. R. et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 363, eaar7785 (2019).
Mancabelli, L. et al. Guideline for the analysis of the microbial communities of the human upper airways. J. Oral. Microbiol. 14, 2103282 (2022).
Kline, M. C., Romsos, E. L. & Duewer, D. L. Evaluating digital PCR for the quantification of human genomic DNA: accessible amplifiable targets. Anal. Chem. 88, 2132–2139 (2016).
Zhang, X., Osaka, T. & Tsuneda, S. Bacterial metabolites directly modulate farnesoid X receptor activity. Nutr. Metab. 12, 48 (2015).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Shaiber, A. et al. Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. Genome Biol. 21, 292 (2020).
Koster, J. & Rahmann, S. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012).
Eren, A. M. et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 (2015).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
Eren, A. M., Vineis, J. H., Morrison, H. G. & Sogin, M. L. A filtering method to generate high quality short reads using illumina paired-end technology. PLoS ONE 8, e66643 (2013).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Lee, M. D. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35, 4162–4164 (2019).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput Biol. 7, e1002195 (2011).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Katz, K. S. et al. STAT: a fast, scalable, MinHash-based k-mer tool to assess Sequence Read Archive next-generation sequence submissions. Genome Biol. 22, 270 (2021).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Acknowledgements
We acknowledge assistance with animal experiments from Caltech Office of Laboratory Animal Research. We thank M. Ratanapanichkich (California Institute of Technology) for assistance on manual refinement of metagenomic bins and feedback on figure design. We thank A. Carter (California Institute of Technology) for assistance with Quant-seq library preparation, ddPCR measurements and feedback during manuscript preparation. We thank M. Cooper (California Institute of Technology) for identifying appropriate statistical tests, guidance during Quant-seq analysis and feedback on figure design. We thank S. R. Bogatyrev for preliminary investigations, discussions and advice. We thank O. Pradhan (California Institute of Technology) and R. Akana (California Institute of Technology) for advice and feedback during manuscript preparation. We thank B. McDonald (University of Chicago) for providing his expertise and advice on clinical sample collection and processing. We thank A. Wang (University of Chicago) for her assistance in the processing of the human tissue for Figs. 3–6. We thank N. Shelby (California Institute of Technology) for contributions to writing and editing this manuscript. This work was funded in part by a grant from the Kenneth Rainin Foundation (grant no. 2018-1207 to R.F.I.), the Army Research Office Multidisciplinary University Research Initiative (grant no. W911NF-17-1-0402 to R.F.I.), the Jacobs Institute for Molecular Engineering for Medicine, a NIH NIDDK grant (no. RC2 DK133947 to R.F.I. and B.J.), a National Science Foundation Graduate Research Fellowship (grant no. DGE‐1745301 to N.J.W.-W.), and a National Institutes of Health Biotechnology Leadership Pre-doctoral Training Program fellowship from Caltech’s Donna and Benjamin M. Rosen Bioengineering Center (grant no. T32GM112592, to J.T.B.), a Helmsley Foundation grant (to F.T.), a NIH NIDDK grant (no. RC2 DK122394, to F.T.), a F30 (grant no. 5F30DK121470, to D.G.S.), a R01 (grant no. DK067180, to B.J.) and the Digestive Diseases Research Core Center grant no. P30 DK42086 at the University of Chicago (to B.J.). The funders had no role in the design of the study, the collection, analysis and interpretation of data, nor in writing the manuscript.
Author information
Authors and Affiliations
Contributions
N.J.W.-W. and J.T.B. conceived and optimized MEM. J.T.B. designed sample collection and analyzed 16S sequencing. D.G.S. codesigned and performed human biopsy collection. N.J.W.-W. and F.T. analyzed shotgun sequencing. A.E.R. performed library preparation. R.F.I. contributed to the design and implementation of the study and to obtaining funding. A.M.E. oversaw the bioinformatic analysis, contributed to the design and implementation of the study and to obtaining funding. B.J. supervised the clinical work, contributed to the design and implementation of the study and to obtaining funding. All authors edited the manuscript. A detailed author contribution statement is available in the Supplementary Information.
Corresponding author
Ethics declarations
Competing interests
The work in this paper is the subject of a patent application filed by Caltech (R.F.I., N.J.W.-W., J.T.B. and A.E.R.). The other authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Lei Tang and Hui Hua, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Correlation between bacterial load and non-host reads.
Shotgun sequencing was performed on longitudinally sampled intestinal biopsies after processing with host depletion (N = 60 biological replicates). Roughly 25 million reads on average were obtained for each biopsy and all samples fit on a single NovaSeq S1 flowcell. After host-filtering an average of 2 million reads were remaining with a range from 2E4 reads to 2E7 reads. For each box, the middle horizontal line denotes mean values, boxes extend to the 25th and 75th percentile, and whiskers extend to the 1.5 interquartile range. The variability in non-host reads remaining had a strong correlation (Spearman, r = 0.79) with the total microbial load as measured by digital PCR. This strong correlation indicated that our process was achieving a relatively uniform depletion across all samples. Additionally, the strong correlation indicates that the majority of non-human reads in our samples come from bacteria picked up by the 16S primers used for total microbial load quantification.
Extended Data Fig. 2 Bacterial loads of longitudinal biopsies.
16S rRNA gene copies were quantified as a proxy for bacterial load for all biopsies. Samples were plotted by participant and then by location. (N = 3 biological replicates for each location for each participant, LOB refers to limit of blank defined as LoB = meanblank + 1.645[SDblank] based on three processing blanks).
Extended Data Fig. 3 Longitudinal variation at the pathways and gene-level.
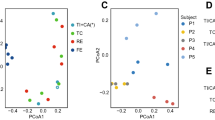
PCA analysis was performed on all 60 longitudinal samples grouped by participant (CT7, CT8, CT12, CT13, and CT14). Shotgun-sequencing data was annotated for pathways and genes through HUMAnN 3 without the taxonomic-profile flag. A) PCA on relative abundance of all pathways. B) PCA on relative abundance of completed pathways (defined as above 90% of modules being present). C) PCA on relative abundance of all genes. D) PCA on relative abundance of the top 5,000 most abundant genes in each participant.
Extended Data Fig. 4 Archaeon Methanobrevibacter smithii found along the lower GI tract.
From shotgun sequencing, we detected participant CT12 had low levels of Methanobrevibacter smithii present in the terminal ileum, descending colon, and rectal biopsies (N = 3 biological replicates; error bars are 95% CI centered on the mean). MAG construction was performed on co-assembly of all biopsies taken from the terminal ileum and descending colon to reconstruct a full Methanobrevibacter smithii genome (completeness: 100%, redundancy: 0%).
Extended Data Fig. 5 Ruminococcus bromii strain variants at the nucleotide (SNV), codon (SCV), and amino acid (AA) level.
SNVs present in R. bromii above the threshold of 21% deviation from reference were analyzed at the codon and translated-level to determine if SNVs may indicate a functional change. The fixation index for each level of analysis were plotted.
Supplementary information
Supplementary Information
Supplementary Figs. 1–9 and Author Contact Information/ORCID.
Supplementary Tables 1–11
A description for each table can be found as a note in the first cell of each sheet.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu-Woods, N.J., Barlow, J.T., Trigodet, F. et al. Microbial-enrichment method enables high-throughput metagenomic characterization from host-rich samples. Nat Methods 20, 1672–1682 (2023). https://doi.org/10.1038/s41592-023-02025-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-023-02025-4
