
Abstract
The cost of maintaining exabytes of data produced by sequencing experiments every year has become a major issue in today’s genomic research. In spite of the increasing popularity of third-generation sequencing, the existing algorithms for compressing long reads exhibit a minor advantage over the general-purpose gzip. We present CoLoRd, an algorithm able to reduce the size of third-generation sequencing data by an order of magnitude without affecting the accuracy of downstream analyses.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

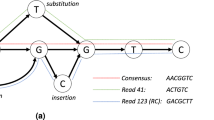
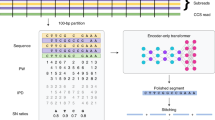
Data availability
ONT datasets: Mycoplasma bovis24, ERR4179765; M. bovis Bonito24, ERR4179766; vir25, ERR2708427–ERR2708436; lun26, PRJEB30781; Sorghum27, SRX4104135–SRX4104138; Zymo2, https://nanopore.s3.climb.ac.uk/Zymo-GridION-EVEN-BB-SN.fq.gz; Zymo2 R10, https://s3.climb.ac.uk/nanopore/Zymo-GridION-EVEN-BB-SN-PCR-R10HC-flipflop.fq.gz; Zymo2 Bonito, ERR5396170; Zymo2 PromethION, https://nanopore.s3.climb.ac.uk/Zymo-PromethION-EVEN-BB-SN.fq.gz; NA12878 (ref. 1), http://s3.amazonaws.com/nanopore-human-wgs/rel6/rel_6.fastq.gz; CHM13, https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/nanopore/rel6/rel6.fastq.gz; CHM13 Bonito, https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/nanopore/rel7/rel7.fastq.gz. PacBio HiFi datasets: mosquito, SRX8642991, SRX8642992; Drosophila28, SRX499318; strawberry29, SRR11606867; ATCC WGS, PRJNA546278; mouse29, SRR11606870; HG002 HiFi30, SRR10382244, SRR10382245, SRR10382248, SRR10382249; CHM13 HiFi, SRR11292120–SRR11292123. PacBio CLR datasets: yeast, http://gembox.cbcb.umd.edu/mhap/raw/yeast_filtered.fastq.gz; Arabidopsis, http://gembox.cbcb.umd.edu/mhap/raw/athal_filtered.fastq.gz; Macadamia31, SRR11191909; 48 plex, https://downloads.pacbcloud.com/public/dataset/MicrobialMultiplexing_48plex/48-plex_sequences/; CHM1, http://datasets.pacb.com/2014/Human54x/fastq.html; HG002 CLR32, SRX5590586. Other: CHM13 T2T assembly version 1.1 (ref. 16), GCA_009914755.3; GRCh38 assembly, GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz; Sorghum assembly, GCF_000003195.3; M. bovis assembly, GCF_000183385.1; Genome in a Bottle version 4.2.1 HG002 reference variants20, HG002_GRCh38_1_22_v4.2.1_benchmark.vcf.gz.
Code availability
Source code for CoLoRd is available at https://github.com/refresh-bio/colord. The package can be installed via Bioconda at https://anaconda.org/bioconda/colord.
References
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
Stancu, M. C. et al. Mapping and phasing of structural variation in patient genomes using Nanopore sequencing. Nat. Commun. 8, 1326 (2017).
Jones, D. C., Ruzzo, W. L., Peng, X. & Katze, M. G. Compression of next-generation sequencing reads aided by highly efficient de novo assembly. Nucleic Acids Res. 40, e171 (2012).
Bonfield, J. K. & Mahoney, M. V. Compression of FASTQ and SAM format sequencing data. PloS ONE 8, e59190 (2013).
Roguski, Ł. & Deorowicz, S. DSRC 2: industry-oriented compression of FASTQ files. Bioinformatics 30, 2213–2215 (2014).
Grabowski., S., Deorowicz, S. & Roguski, Ł. Disk-based compression of data from genome sequencing. Bioinformatics 31, 1389–1395 (2015).
Roguski, Ł., Ochoa, I., Hernaez, M. & Deorowicz, S. FaStore: a space-saving solution for raw sequencing data. Bioinformatics 34, 2748–2756 (2018).
Liu, Y., Yu, Z., Dinger, M. E. & Li, J. Index suffix–prefix overlaps by (w, k) -minimizer to generate long contigs for reads compression. Bioinformatics 35, 2066–2074 (2018).
Chandak, S., Tatwawadi, K., Ochoa, I., Hernaez, M. & Weissman, T. SPRING: a next-generation compressor for FASTQ data. Bioinformatics 35, 2674–2676 (2018).
Dufort y Álvarez., G. et al. ENANO: encoder for NANOpore FASTQ files. Bioinformatics 36, 4506–4507 (2020).
Nicolae, M., Pathak, S. & Rajasekaran, S. LFQC: a lossless compression algorithm for FASTQ files. Bioinformatics 31, 3276–3281 (2015).
Myers, E. The fragment assembly string graph. Bioinformatics 21, 79–85 (2005).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016).
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R. & Phillippy, A. M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Dufort y Álvarez., G. et al. RENANO: a REference-based compressor for NANOpore FASTQ files. Bioinformatics 37, 4862–4864 (2021).
Nurk, S. et al. The complete sequence of a human genome. Preprint at bioRxiv https://doi.org/10.1101/2021.05.26.445798v1 (2021).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Zook, J. M. et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 37, 561–566 (2019).
Deorowicz, S. FQSqueezer: k-mer-based compression of sequencing data. Sci. Rep. 10, 578 (2020).
Kokot, M., Długosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761 (2017).
Sosić, M. & Sikić, M. Edlib: a C/C++ library for fast, exact sequence alignment using edit distance. Bioinformatics 33, 1394–1395 (2017).
Vereecke, N. et al. High quality genome assemblies of Mycoplasma bovis using a taxon-specific Bonito basecaller for MinION and Flongle long-read Nanopore sequencing. BMC Bioinformatics 21, 517 (2020).
Depledge, D. P. et al. Direct RNA sequencing on Nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 10, 754 (2019).
Charalampous, T. et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 7, 783–792 (2019).
Deschamps, S. et al. A chromosome-scale assembly of the sorghum genome using Nanopore sequencing and optical mapping. Nat. Commun. 9, 4844 (2018).
Kim, K. et al. Long-read, whole-genome shotgun sequence data for five model organisms. Sci. Data 1, 140045 (2014).
Hon, T. et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 7, 399 (2020).
Cheng, H., Concepcion, G. T., Feng, X. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Murigneux, V. et al. Comparison of long-read methods for sequencing and assembly of a plant genome. GigaScience 9, giaa146 (2020).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162 (2019).
Acknowledgements
This work was supported by the National Science Centre, Poland, project DEC-2019/33/B/ST6/02040 (M.K., A.G. and S.D.) and by the US National Institutes of Health (grants R01HG010040, U01HG010971 and U41HG010972 to H.L.).
Author information
Authors and Affiliations
Contributions
M.K. and S.D. designed and implemented the algorithm. H.L. designed the variant-calling and consensus analyses, investigated and described their results. A.G., S.D. and M.K. conducted experiments. A.G. prepared visualizations and wrote the majority of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
H.L. is a consultant of Integrated DNA Technologies and is on the scientific advisory boards of Sentieon and Innozeen. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Lin Tang, in collaboration with the Nature Methods team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Tables 1–4 and Fig. 1
Supplementary Data
Detailed results: compression, consensus generation and variant calling.
Rights and permissions
About this article

Cite this article
Kokot, M., Gudyś, A., Li, H. et al. CoLoRd: compressing long reads. Nat Methods 19, 441–444 (2022). https://doi.org/10.1038/s41592-022-01432-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-022-01432-3
This article is cited by
-
PMFFRC: a large-scale genomic short reads compression optimizer via memory modeling and redundant clustering
BMC Bioinformatics (2023)
-
Reference-free lossless compression of nanopore sequencing reads using an approximate assembly approach
Scientific Reports (2023)
