
Abstract
Cryogenic electron microscopy (cryo-EM) maps are now at the point where resolvability of individual atoms can be achieved. However, resolvability is not necessarily uniform throughout the map. We introduce a quantitative parameter to characterize the resolvability of individual atoms in cryo-EM maps, the map Q-score. Q-scores can be calculated for atoms in proteins, nucleic acids, water, ligands and other solvent atoms, using models fitted to or derived from cryo-EM maps. Q-scores can also be averaged to represent larger features such as entire residues and nucleotides. Averaged over entire models, Q-scores correlate very well with the estimated resolution of cryo-EM maps for both protein and RNA. Assuming the models they are calculated from are well fitted to the map, Q-scores can be used as a measure of resolvability in cryo-EM maps at various scales, from entire macromolecules down to individual atoms. Q-score analysis of multiple cryo-EM maps of the same proteins derived from different laboratories confirms the reproducibility of structural features from side chains down to water and ion atoms.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
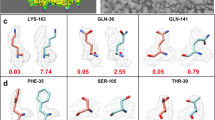
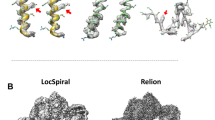

Data availability
The cryo-EM maps of apoferritin have been deposited in the EMDB with accession codes 20026 (1.75 Å), 20027 (2.3 Å) and 20028 (3.1 Å). The figures show these maps and also other maps and models available in the EMDB and PDB (accession codes specified in the figure captions, see also Supplementary Tables 1 and 2). All data including calculations based on these maps that support the findings of this study are available from the corresponding author upon request.
Code availability
Q-scores are implemented in the MapQ plugin to UCSF Chimera and available on GitHub and Zenodo35. A tutorial is also available at the link under the ‘tutorials’ folder. Pseudo code is provided under the ‘docs’ folder.
References
Anonymous. Challenges for cryo-EM. Nat. Methods 15, 985–985 (2018).
Henderson, R. et al. Outcome of the first electron microscopy validation task force meeting. Struct. Lond. Engl. 1993 20, 205–214 (2012).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Heymann, J. B. Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. Publ. Protein Soc. 27, 159–171 (2018).
Anonymous. Local resolution of cryo-EM maps with MonoRes. Nat. Methods 15, 246 (2018).
Pintilie, G. & Chiu, W. Comparison of Segger and other methods for segmentation and rigid-body docking of molecular components in cryo-EM density maps. Biopolymers 97, 742–760 (2012).
Terwilliger, T. C., Adams, P. D., Afonine, P. V. & Sobolev, O. V. A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps. Nat. Methods 15, 905 (2018).
Joseph, A. P., Lagerstedt, I., Patwardhan, A., Topf, M. & Winn, M. Improved metrics for comparing structures of macromolecular assemblies determined by 3D electron-microscopy. J. Struct. Biol. 199, 12–26 (2017).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D. 74, 531–544 (2018).
Trabuco, L. G. et al. Applications of the molecular dynamics flexible fitting method. J. Struct. Biol. 173, 420–427 (2011).
Joseph, A. P. et al. Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment. Methods 100, 42–49 (2016).
Pintilie, G., Chen, D.-H., Haase-Pettingell, C. A., King, J. A. & Chiu, W. Resolution and probabilistic models of components in cryoEM maps of mature P22 bacteriophage. Biophys. J. 110, 827–839 (2016).
DiMaio, F., Zhang, J., Chiu, W. & Baker, D. Cryo-EM model validation using independent map reconstructions. Protein Sci. 22, 865–868 (2013).
Trabuco, L. G., Villa, E., Schreiner, E., Harrison, C. B. & Schulten, K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods 49, 174–180 (2009).
DiMaio, F., Tyka, M. D., Baker, M. L., Chiu, W. & Baker, D. Refinement of protein structures into low-resolution density maps using rosetta. J. Mol. Biol. 392, 181–190 (2009).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Pintilie, G. & Chiu, W. Assessment of structural features in Cryo-EM density maps using SSE and side chain Z-scores. J. Struct. Biol. 204, 564–571 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Dunitz, J. D., Schomaker, V. & Trueblood, K. N. Interpretation of atomic displacement parameters from diffraction studies of crystals. J. Phys. Chem. 92, 856–867 (1988).
Trueblood, K. N. et al. Atomic dispacement parameter nomenclature. Report of a subcommittee on atomic displacement parameter nomenclature. Acta Crystallogr. A. 52, 770–781 (1996).
Carugo, O. How large B-factors can be in protein crystal structures. BMC Bioinformatics 19, https://doi.org/10.1186/s12859-018-2083-8 (2018).
Hyrc, C. F. et al. Accurate model annotation of a near-atomic resolution cryo-EM map. Proc. Natl Acad. Sci. USA 114, 3103–3108 (2017).
Herzik, M. A., Fraser, J. S. & Lander, G. C. A multi-model approach to assessing local and global cryo-EM map quality. Struct. Lond. Engl. 1993 27, 344–358.e3 (2019).
Singharoy, A. et al. Molecular dynamics-based refinement and validation for sub-5 Å cryo-electron microscopy maps. eLife 5, e16105 (2016).
Meyder, A., Nittinger, E., Lange, G., Klein, R. & Rarey, M. Estimating electron density support for individual atoms and molecular fragments in X-ray structures. J. Chem. Inf. Model. 57, 2437–2447 (2017).
Wahab, M. A. Essentials of Crystallography (Alpha Science International, 2009).
Graef, M. D. Introduction to Conventional Transmission Electron Microscopy (Cambridge Univ. Press, 2003).
Holton, J. M. A beginner’s guide to radiation damage. J. Synchrotron Radiat. 16, 133–142 (2009).
Darby, J. F. et al. Water networks can determine the affinity of ligand binding to proteins. J. Am. Chem. Soc. 141, 15818–15826 (2019).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. 66, 12–21 (2010).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Bell, J. M., Chen, M., Durmaz, T., Fluty, A. C. & Ludtke, S. J. New software tools in EMAN2 inspired by EMDatabank map challenge. J. Struct. Biol. 204, 283–290 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Pintilie, G. MapQ (GitHub, 2019); https://github.com/gregdp/mapq
Acknowledgements
This research has been supported by National Institutes of Health grants (nos. R01GM079429, P41GM103832 and S10OD021600) to W.C. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco (supported by grant no. NIGMS P41GM103311). We thank F. Sun and X.J. Huang (Institute of Biophysics, CAS) for the human apoferritin samples. We also thank C. Lawson for discussions related to B-factors and ADPs.
Author information
Authors and Affiliations
Contributions
G.P. and W.C. conceived Q-scores. G.P. implemented the software and performed testing. K.Z. collected the images and reconstructed the maps (EMDB 20026, 20027 and 20028). Z.S. and S.L. provided additional data (not shown) for testing the Q-score. M.F.S. and W.C. contributed the discussion during the development. G.P. wrote the manuscript with inputs from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Average Q-scores are low when the model is not well-fitted to the map.
(a)A local rotational search around a segmented Apoferritin map (EMD:20026, 1.75Å). The top 10 fits by cross-correlation (CC) score are considered, out of 100 fits with evenly sampled orientations around the protein center. The plot, top left, shows CC and Q-scores plotted for each of the 10 top fits. CC and Q-scores are both lower for incorrect placements of the model. (b) Cross-correlation scores before and after real-space refinement with Phenix – the scores increase after refinement in all cases. (c) Q-scores also increase in all cases after refinement. The maps and models for which Q-scores are calculated are listed in Table 1.
Supplementary Figure 2 Q-scores for atoms in X-ray maps with resolutions of 0.48Å to 1.30Å.
In each plot, the curve labeled u is the atomic profile extracted from each map, and v is the curve corresponding to the reference Gaussian with width of 0.6Å. At increasing resolutions, atomic profiles become narrower compared to the reference Gaussian, and Q-scores decrease.
Supplementary Figure 3 The effect of using different parameters for the reference gaussian on the Q-score.
In (i) and (ii), the height of the reference gaussian (RG) is 10𝜎M and 1𝜎M, respectively, above the mean avgM of all values in the map. With normalized cross-correlation (CC), using the shorter RG results in a lower score, as shown in the graph at the bottom. However, with cross-correlation about the mean (CCm), the score is the same in both cases. Adding an offset to both the RG and atom profile (iii), the CC score increases, whereas the CCm stays the same. Panels (iv, v and vi), repeat the same but with larger width for the RG (𝜎=1.0). As before, CCm scores stay the same across different conditions, while CC scores vary when using different RG height and offset. All atom profiles come from the map of Apoferritin at 1.75Å resolution (EMD:20026), atom CD2 in Leu26.
Supplementary Figure 4 Model Q-score vs. map grid step.
The map shown is Apoferritin 3.1Å resolution, EMD:20028. The original map has a step size of 0.6Å. New maps were generated by resampling this map, using tri-linear interpolation, on new grids which cover the same volume but have larger step sizes. The position of all grid points other than the origin (0,0,0) change as a result of this process. Q-scores calculated for the same model (PDB:3ajo, 1473 protein atoms) are very similar for step sizes of 0.6 to 1.2, but start to decrease beyond this point as the step size becomes too large for the resolution of the map (3.1Å).
Supplementary Figure 5 Effect of sharpening on Q-score.
The original map (GroEL map entry 104 in 2016 EMDB map challenge) with reported resolution of 4.4Å by gold-standard FSC. The Q-score is calculated using a fitted model (PDB:4hel, 3864 atoms in chain A). Q-scores increase with sharpening (using phenix.auto_sharpen b_sharpen=B_sharpen) up to 0.31 with B_sharpen of 275, and then start to decrease with further sharpening. Visually the map has the most detail after sharpening with B_sharpen of 275, corresponding to the highest Q-score, past which it starts to become fragmented with more sharpening.
Supplementary Figure 6 Q-scores for cryoEM maps and models in EMDB (blue, also shown in Figure 5) and from simulated maps (orange).
(a) Average Q-scores for atoms in protein components. The simulated maps were generated with phenix.fmodel scattering_table=electron followed by phenix.mtz2map, applied to a single protein from the Apoferritin model PDB:3ajo (1473 atoms). (b) Average Q-scores for atoms in nucleic acid components. Simulated maps were generated same as in (A) but for chain 1, residues 883-912, from PDB:6az3 (656 atoms).
Supplementary Figure 7 Plots of average B-factors vs. resolution for two sets of structures in the PDB.
(a, c) Structures up to Sep. 2018. (b, d) All structures between Jan. 2015 – Sep. 2018. Each point corresponds to one structure, for which the average B-factor is plotted vs. resolution. In (A,B), a 2nd degree polynomial is fit to the points; in (C,D), a log function is fit to the points; the r2 is similar in both cases. The data for the plots was obtained from the PDB website (https://www.rcsb.org/) using Advanced Search. The PDB ids used in the search were obtained from a prior analysis1, which include PDB ids up to Sep. 2018. 1. Shao, C., Liu, Z., Yang, H., Wang, S. & Burley, S. K. Outlier analyses of the Protein Data Bank archive using a probability-density-ranking approach. Sci. Data 5, 180293 (2018).
Supplementary Figure 8 B-factors vs. Q-scores.
B-factors (as deposited with the PDB structures) vs. Q-scores (calculated in a cryoEM maps at similar resolutions). Each point represents an atom in the model. Note that the Q-score axes are inverted (-1 to 1) in order to allow a log-curve to be fitted in (C,D). The r2 are slightly better for log relationships. Two models are used: PDB:3ajo (1473 protein atoms) and PDB: 3wnw (1433 protein atoms).
Supplementary Figure 9 ADPs vs Q-scores.
Correlation between atomic displacement parameters (ADPs) and Q-scores, both calculated in a CryoEM map of Apoferritin, EMD:9865 at 1.54Å resolution, using a rigidly fitted model (PDB:3ajo). Each point represents an atom in the model. Note that the Q-score axes are inverted (-1 to 1) in order to allow log curves to be fitted in C,D. ADPs were calculated with Phenix in real space (phenix.real_space_refine run=adp) (A,C) and reciprocal space (with phenix.refine) (B,D). ADPs were also calculated without restraints which are applied by default, however lower correlations were obtained (not shown).
Supplementary Figure 10 Radial plots of distances from water and Mg atoms to other atoms.
Distribution of Q-scores for solvent atoms (229 water and 12 Mg) in (a) X-ray map (PDB:3ajo), and in (b, c) two cryoEM maps. In (B) and (C), scores are shown before and after refinement.
Supplementary Figure 11 Radial plots of distances from water and Mg atoms to other atoms.
Oxygen atoms in water are labeled H2O, whereas oxygen/nitrogen atoms in protein are labeled O/N. In the X-ray structure (a), sharp peaks can be seen at 2.8Å for H2O-H2O (water-water) and H2O-O (water-protein). In models fitted and refined in cryoEM maps (b, c), a H2O-O peak can also be seen at 2.8Å, however peaks for H2O-H2O (‘second-layer’ water) are more broad/diffuse.
Supplementary information
Supplementary Information
Supplementary Figs. 1–11 and Tables 1 and 2.
Source data
Rights and permissions
About this article

Cite this article
Pintilie, G., Zhang, K., Su, Z. et al. Measurement of atom resolvability in cryo-EM maps with Q-scores. Nat Methods 17, 328–334 (2020). https://doi.org/10.1038/s41592-020-0731-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-020-0731-1
This article is cited by
-
Dynamic inter-domain transformations mediate the allosteric regulation of human 5, 10-methylenetetrahydrofolate reductase
Nature Communications (2024)
-
Substrate binding plasticity revealed by Cryo-EM structures of SLC26A2
Nature Communications (2024)
-
Structural basis of prostaglandin efflux by MRP4
Nature Structural & Molecular Biology (2024)
-
Automated model building and protein identification in cryo-EM maps
Nature (2024)
-
Platform-directed allostery and quaternary structure dynamics of SAMHD1 catalysis
Nature Communications (2024)
