
Abstract
Treatment with immune checkpoint blockade (ICB) frequently triggers immune-related adverse events (irAEs), causing considerable morbidity. In 214 patients receiving ICB for melanoma, we observed increased severe irAE risk in minor allele carriers of rs16906115, intronic to IL7. We found that rs16906115 forms a B cell-specific expression quantitative trait locus (eQTL) to IL7 in patients. Patients carrying the risk allele demonstrate increased pre-treatment B cell IL7 expression, which independently associates with irAE risk, divergent immunoglobulin expression and more B cell receptor mutations. Consistent with the role of IL-7 in T cell development, risk allele carriers have distinct ICB-induced CD8+ T cell subset responses, skewing of T cell clonality and greater proportional repertoire occupancy by large clones. Finally, analysis of TCGA data suggests that risk allele carriers independently have improved melanoma survival. These observations highlight key roles for B cells and IL-7 in both ICB response and toxicity and clinical outcomes in melanoma.
Similar content being viewed by others

Main
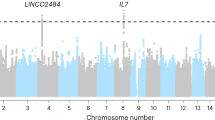
Immune checkpoint blockade (ICB), consisting of monoclonal antibodies targeted against the proteins PD-1 and CTLA-4, can elicit durable remission of metastatic melanoma in a subset of individuals1,2,3 but is associated with autoimmune complications (immune-related adverse events (irAEs)), for which risk factors are poorly characterized4,5,6. Both oncological outcomes and occurrence of irAEs after ICB treatment vary markedly between patients1,7. We postulate that irAE development can be influenced by cancer-independent factors, including germline genetics. A parallel genome-wide association study (GWAS) identified carriage of the minor allele of rs16906115, a non-coding polymorphism at IL7, to be associated with increased risk of ICB-associated irAEs. In the present study, we replicated this clinical association in a cohort of 214 patients receiving ICB for melanoma, finding that carriers of the minor allele of rs16906115 (referred to as the risk allele) have significantly increased risk of toxicities requiring corticosteroid treatment. RNA sequencing (RNA-seq) performed on isolated immune cell types from patients pre-ICB and post-ICB treatment (n = 194), and healthy controls (n = 170), demonstrates markedly increased IL7 expression in patient-derived B cells before treatment, with this effect being significantly greater in risk allele carriers. Notably, independent of genotype, we found that increased B cell IL7 expression pre-treatment is associated with risk of irAEs, indicative of causality. By performing single-cell (sc) RNA-seq of B cells from patients and controls, we delineated B cell subsets expressing IL7 and note that risk allele carriage is associated with increased B cell maturation. Finally, reflecting the importance of IL-7 in T cell maturation, we found genotype-specific T cell receptor (TCR) repertoire skewing with increased proportional repertoire occupancy by hyper-expanded CD8+ T cell clones in response to ICB treatment. Separate analysis of The Cancer Genome Atlas (TCGA) data demonstrates that the risk allele is associated with disease-specific and overall survival in melanoma, indicating that this pathway plays a role in the natural history of melanoma progression. In summary, we replicated and mechanistically dissected the effects of the leading identified risk locus for ICB-induced irAEs, implicating a key role for B cells and IL-7 in mediation of immunological responses and toxicity to these therapies.
Results
rs16906115 is a risk allele for irAE development post-ICB
We sought to replicate the reported association between rs16906115 and irAE development in a dataset of 214 prospectively recruited and genotyped patients who had received ICB for melanoma (Extended Data Fig. 1a), in which rs16906115 had a minor allele frequency (MAF) of 7.4%. We found that carriage of the minor risk (A) allele (one or two copies) was associated with an increased likelihood of developing severe (grade 3 or higher) irAEs requiring corticosteroids before the fifth cycle (C5) of treatment across all patients, with an odds ratio (OR) of 2.24 (95% confidence interval (CI): 1.03–5.09, P = 0.046) (Fig. 1a). In the primary irAE GWAS dataset, 90% of patients were treated with single-agent anti-PD-1 (sICB) or anti-PDL1, as opposed to combination anti-CTLA-4/anti-PD-1 (cICB). Given that sICB recipients have only ~20% the risk of cICB recipients for developing irAEs1,3,7, the sensitivity to detect genetic effects is likely greater with this more moderate treatment. In keeping with this, we found that sICB (anti-PD-1) recipients carrying the risk allele had a 6.0 OR (CI: 1.5–23.0, P = 0.0084) of developing irAEs requiring steroids before C5, as well as increased risk of requiring steroids at any timepoint on treatment (OR 4.4, 95% CI: 1.2–15.1, P = 0.019) (Fig. 1a). The single-nucleotide polymorphism (SNP) rs16906115 is intronic to IL7, encoding the cytokine IL-7, a key lymphopoietic cytokine with pleiotropic effects across lymphocyte subsets8,9,10. We, therefore, explored the relationship between rs16906115 status and ICB-induced changes in lymphocyte count, as measured at routine hospital blood tests. Comparison of pre-treatment lymphocyte count with the first count measured at least 21 days after treatment initiation (referred to as lymphocyte stability (LS)) revealed that, although cICB treatment did not significantly affect LS, sICB treatment elicited a fall in LS (Extended Data Fig. 1b), which was consistent across recipients of nivolumab and pembrolizumab (Extended Data Fig. 1c). In particular, non-carriers of the risk allele had significantly reduced LS post-sICB (median pre-treatment value 1.82 × 109 cells per liter, interquartile range (IQR) 1.26–2.33; post-treatment value 1.52 × 109 cells per liter, IQR 1.07–2.11) (P = 0.00044; Fig. 1b), a reduction not observed in risk allele carriers (median pre-treatment value 1.35 × 109 cells per liter, IQR 1.11–2.72; post-treatment 1.53 × 109 cells per liter, IQR 1.34–1.90) (P = 0.68; Fig. 1b). Correspondingly, rs16906115 status was associated with the magnitude of sICB-induced change in lymphocyte count (non-carriers: −0.15 × 109 cells per liter; IQR −0.38 to 0.02 versus carriers: −0.025 × 109 cells per liter; IQR −0.11 to 0.36) (P = 0.023; Fig. 1c). Notably, individuals with a fall in LS >20% had reduced progression-free and overall survival (Extended Data Fig. 1d,e), reinforcing the clinical importance of LS and demonstrating an association between rs16906115 and post-treatment lymphopoiesis, in keeping with the known properties of IL-7.

a, OR for developing irAEs requiring steroids before C5 of treatment, or ever, according to allele carriage of rs16906115 and treatment type, and all ICB. Whiskers represent 95% CI; two-sided Fisher exact test, n = 214 patients (98 sICB and 116 cICB). b, Pre-treatment versus first post-treatment lymphocyte count for patients receiving sICB, split by allele; two-sided Wilcoxon signed-rank test, n = 60 patients. c, Difference between pre-treatment and post-sICB treatment lymphocyte count by allele; one-sided Student’s t-test, n = 60 patients.
rs16906115 regulates B cell IL7 expression in patients with melanoma
Plasma IL-7 concentration forms a rate-limiting determinant of T cell expansion, being regulated, in part, by peripheral lymphocyte consumption, including activated T cells11. In keeping with the high levels of peripheral T cell mitosis in melanoma12,13,14, plasma IL-7 levels were undetectable by ELISAs in patient samples. We, therefore, explored IL7 expression across immune subsets isolated from patients and healthy individuals. Although circulating immune cells are not generally recognized to produce IL-7 (refs. 11,15,16,17), we found that peripheral blood isolated CD19+ B cells robustly express IL7 at levels greatly in excess of other cell subsets. Notably, IL7 expression in pre-treatment patient-derived B cells was significantly induced compared to that in B cells from healthy controls (94 controls and 92 patients; mean log2 expression 8.30 FKPM versus 8.87 FKPM, P = 5.9 × 10−6), with this effect not apparent across other subsets and independent of age (Fig. 2a and Extended Data Fig. 2a). This upregulation persisted in early post-ICB treatment samples, with neither ICB type affecting B cell IL7 expression at day 21 (Extended Data Fig. 2b). Crucially, we found that patient risk allele carriers at rs16906115 demonstrated increased B cell IL7 induction versus non-carriers both pre-treatment and over the course of treatment, but genetic effects were not observed across other cell types in patients (Fig. 2b and Extended Data Fig. 2c). Conversely, genetic effects were not observed in B cells or other cell types in healthy individuals (Extended Data Fig. 2d), indicating that rs16906115 forms a B cell cis expression quantitative trait locus (eQTL) in the context of melanoma-associated immune activity. We did not identify genotype-associated B cell IL7 splice variation but, instead, observed upregulation of the main IL7 transcripts, indicating activity through modulation of total IL7 gene expression (Extended Data Fig. 2e). Finally, using flow cytometry, we were able to consistently detect an IL-7+ population of B cells in peripheral blood mononuclear cells (PBMCs), with no IL-7+ cells detected in other major lineages (Extended Data Fig. 2f,g). To further assess evidence for B cell IL7 directly mediating the risk effect of rs16906115 on irAE development, we tested association using random effects mixed linear models18. Consistent with B cell IL7 mediating the genotypic association, we found that, when controlling for ICB type and rs16906115 status, pre-treatment B cell IL7 was independently associated with irAE risk (P = 0.0092), whereas, when controlling for B cell IL7 and ICB type, no genotypic effect was apparent (Fig. 2c). This remained the case when restricting analysis to individuals carrying the reference allele (P = 0.0087), underlying the importance of B cell IL7 to irAE development and providing mechanistic insight into rs16906155 functionality.

a, Expression of IL7 measured using RNA-seq according to status (patient samples are untreated; healthy donor (HD)) split by cell type; P value from two-sided ANOVA, controlling for age (n refers to sample size for each cell type by HD or patient). b, Expression of IL7 by allelic carriage and patient status; patient samples represent untreated (C1, n = 91), after the second cycle of treatment (C2, n = 68), the earliest available later sample (late, n = 39) or HDs (n = 92); one-sided Wilcoxon rank-sum test. For box plots in a,b, the central line reflects the median; the box corresponds to 25–75% quartiles; the upper whisker extends to the largest value no farther than 1.5× IQR; and the lower whisker extends from the 25% quartile to the smallest value no farther than 1.5× IQR. c, Results from linear mixed model random effect analysis; center point shows the estimate for the variable in association with irAE development when controlling for the other two variables; ANOVA using the Satterthwaite method; error bars mark the 95% CI; n = 91 patients.
B cell IL7 expression is associated with maturation
Maturation of B cells is influenced by IL-7 (refs. 9,10), and so we explored the relationship between B cell development and IL7 expression. Analysis of B cells from healthy donors demonstrated correlation of IL7 expression with 11,305 transcripts (adjusted P < 0.05), including the key B cell transcription factors IKZF3 (ref. 19) and POU2AF1 (refs. 20,21) (Fig. 3a and Supplementary Table 2), with remarkably similar results observed in untreated patients (Supplementary Fig. 1c). Pathway analysis of B cell IL7-associated genes using Gene Ontology Biological Process (GOBP) datasets22 demonstrated enrichment in pathways associated with B cell maturation, including ‘immunoglobulin production’ and ‘positive regulation of B cell activation’, whereas anti-correlated genes were associated with TLR4 signaling, neutrophil degranulation and antigen presentation (Fig. 3b, Extended Data Fig. 3a and Supplementary Table 3). Analysis of patient-derived B cells revealed that IL7 expression was anti-correlated with expression of the naive immunoglobulin IGHD but was associated with the percentage of immunoglobulin chains carrying secondary divergent mutations, as observed in somatic hypermutation (Extended Data Fig. 3b). Notably, in keeping with IL7 expression driving these effects, we noted genotypic association of rs16906115 status, with risk allele carriers demonstrating an increased percentage of mutations in IGH, IGK and IGL chains in pre-treated patients (Fig. 3c). Similarly, risk allele carriers showed reduced numbers of IGHD-expressing B cell clones pre-treatment (Fig. 3d), indicating a pre-existent state of B cell immunoglobulin class switching and reduced B cell naivety in these individuals. To further explore IL7 expression and genotypic association of rs16906115 across B cell subsets, we generated scRNA-seq data from B cells (n = 15,755 cells) from pre-treatment patients with metastatic melanoma (n = 24) and healthy controls (n = 5). We identified four major clusters of cells consisting of naive B cells, unswitched memory (USM) cells, switched memory (SM) cells and antibody secreting cells (ASCs) (Fig. 3e and Extended Data Fig. 3c,d). Expression of IL7 was highest in the USM and SM clusters, intermediate in the naive cells and lowest in ASCs (Extended Data Fig. 3e,f) and was significantly higher in patient samples compared to healthy controls across all subsets except ASCs (Fig. 3f). Compared to those from healthy donors, patient naive B cells demonstrated reduced expression of IGHD and concomitant upregulation of IGHM (Extended Data Fig. 3g). Although only four of the patient samples carried the risk allele, we again observed increased IL7 expression in rs16906115 risk allele carriers across all B cells (Fig. 3g), although, within subsets, there was no difference in median expression (Extended Data Fig. 3h). Instead, we found that carriage of the risk allele was associated with proportional changes in B cell subset frequency, with the eQTL effect being driven by significantly increased counts of high IL7-expressing USM cells and a trend to fewer naive B cells (Fig. 3h).

a, Volcano plot demonstrating genes correlated with expression of IL7 from RNA-seq data identified using DESEQ2 from 94 healthy donor (HD) samples, two-tailed Wald test. b, GOBP pathway analysis of genes positively correlated with B cell IL7 (FDR < 0.05); x axis demonstrates fold change above background; y axis demonstrates z-score; all points above dotted line FDR < 0.01; one-tailed hypergeometric test. c, Percentage B cell receptor chains mapped from pre-immunotherapy B cell samples with germline divergent mutations dichotomized by carriage of rs16906115 risk allele, faceted by gene; two-sided Wilcoxon rank-sum test, n = 91. d, Percentage of all B cell clones detected expressing unswitched immunoglobulin IGHD in pre-treatment patient samples dichotomized by carriage of rs16906115 risk allele; two-sided Wilcoxon rank-sum test, n = 91. e, UMAP of scRNA-seq B cells from n = 24 pre-treatment patients and n = 5 healthy donors (15,755 cells total), demonstrating four main populations: naive, USM, SM and ASC. f, Relative IL7 expression across each B cell subset dichotomized according to HD or untreated patient (Mel); two-sided Wilcoxon rank-sum test, n = 24 pre-treatment patients, n = 5 HDs. g, Expression of IL7 across all untreated patient B cells in scRNA-seq data dichotomized according to rs16906115 status; two-sided Wilcoxon rank-sum test, n = 24 pre-treatment patients. h, Frequency of B cell subsets in untreated patients from scRNA-seq data dichotomized according to rs16906115 status; two-sided Wilcoxon rank-sum test, n = 24 pre-treatment patients. For box plots in c,d,f–h, the central line reflects the median value; the box corresponds to 25–75% quartiles; the upper whisker extends to the largest value no farther than 1.5× IQR; and the lower whisker extends from the 25% quartile to the smallest value no farther than 1.5× IQR.
rs16906115 and B cell IL7 associate with T cell responses
Given that T cells are central to the ICB anti-cancer response, are implicated in irAE development and are exquisitely sensitive to IL-7 (refs. 10,12,23,24), we explored the association between T cell ICB responses and genotype. Using flow cytometry of paired pre-treatment and post-treatment PBMC samples, carried out blinded to treatment and genotype (n = 54 paired samples obtained pre-treatment and immediately before the second cycle (C2), median 21 days), we observed genotypic divergence in treatment-induced subset changes. Risk allele carriers showed increases in the proportion of terminally differentiated effector memory T cells re-expressing CD45RA (TEMRA) T cells and a reciprocal fall in naive T cell proportion after ICB (Fig. 4a). In keeping with the known increased sensitivity of CD8+ T cells to IL7 (refs. 25,26), we observed the genotype-specific fall in naive T cells to be most significant in the CD8+ subset (Fig. 4a; median fall homozygous reference allele = −0.31%; IQR −2.80% to 2.27% versus median fall in risk allele carriers = −11.2%; IQR −14.35% to −7.45%; P = 0.00017). Furthermore, we explored the relationship between LS and cell subsets in these individuals, finding that LS was correlated with an increase in all T cells and effector memory cells post-ICB and a fall in naive T cells (Extended Data Fig. 4a). We proceeded to explore the effect of rs16906115 on CD8+ T cell gene expression across recipients of ICB. Given the stronger association of genotype with irAEs in sICB recipients and the markedly larger impact of cICB on CD8+ T cell transcriptomics12, which may saturate any genotypic effects, we restricted analysis to samples from sICB recipients. We performed differential expression analysis of CD8+ cell expression profiles obtained from pre-treatment and post-treatment samples from patients (194 samples from 92 patients: 86 pre-treatment and 108 post-treatment), controlling for confounding variables and dichotomizing by carriage of risk allele at rs16906115. We found that genotype was associated with 570 differentially expressed genes (DEGs) (adjusted P < 0.05; Fig. 4b and Supplementary Table 4), with 96% of transcripts displaying the same direction of effect of genotype across each of the three timepoints (days 0, 21 and 63), showing consistent genotypic effects over time. Supporting the flow cytometry observations, we noted genotypic associations with genes associated with divergent stages of T cell differentiation, including PRDM1, implicated in terminal differentiation of CD8+ T cells27. This gene was upregulated in risk allele carriers, whereas these individuals simultaneously had reduced expression of SH3BGRL2, which was found to strongly correlate with the naive marker CD27, most significantly in patient samples (Pinteraction.status < 0.001) (Fig. 4c and Extended Data Fig. 4b). Gene pathway analysis showed that induced genes were enriched across ten pathways (adjusted P < 0.05), broadly encompassing anabolic processes, including ‘regulation of phosphatidylinositol 3-kinase signaling’, ‘activation of MAPK activity’ and ‘transcription from RNA polymerase II promoter’ (Extended Data Fig. 4c). Pathway enrichment for risk-allele-associated repressed genes was more marked, however, incorporating 40 pathways predominantly composed of cell division processes, including ‘mitotic nuclear envelope disassembly’ and ‘mitotic cytokinesis’ (Fig. 4d and Supplementary Table 5). To assess the evidence for B cell IL7 expression mediating the genotypic association of rs16906115 with CD8+ T cell expression, we correlated CD8+ T cell gene expression with B cell IL7 expression from the same blood sample (n = 189 sICB samples). This identified 935 significant DEGs (adjusted P < 0.05; Supplementary Table 6) associated with B cell IL7. Pathway analysis of anti-correlated genes demonstrated a striking enrichment of those involved in cell division (Fig. 4e and Supplementary Table 7), overlapping highly with pathways repressed by carriage of rs16906115 risk allele (Fig. 4f) and, thus, providing further support for mediation of rs16906115 genotypic effects via B cell IL7 expression. In keeping with this, when controlling for rs16906115 status, B cell IL7-repressed pathways remained highly associated with cell division (Extended Data Fig. 4d and Supplementary Table 8). Finally, we performed the same analysis in non-carriers of the risk allele. This approach reduced the size of the cohort and the range of B cell IL7 expression, resulting in fewer DEGs; however, at a false discovery rate (FDR) threshold of 0.2, the pathways repressed strongly reflected those in the full dataset (Extended Data Fig. 4e and Supplementary Table 9). As a further corroboration, we examined allelic associations with previously described mitotic signature scores23, exploring the change in signature upon treatment across each allele. Consistent with our pathway analysis results, we note significantly lower induction of the mitotic signature in risk allele carriers (Fig. 4g).

a, Differences in subset proportion (depicted in facet name) between baseline untreated (C1) and immediately before the second cycle of treatment (C2) as determined by flow cytometry results assessing change in CD8+ T cell subset (depicted in facet name) with ICB treatment according to rs16906115 status; two-sided Wilcoxon rank-sum test, n = 54 patients. b, Volcano plot depicting results for DESEQ2 differential gene expression analysis of CD8+ T cell RNA-seq data, dichotomized by rs16906115 status; n = 194 samples across three cycles (untreated, C2 and C4) of sICB treatment, two-tailed Wald test. c, Example box plots of genes regulated by allele in sICB recipients, faceted by timepoint; two-sided t-test of normalized expression values, n = 86 patients at baseline, n = 69 patients at day 21. d, GOBP pathway analysis of genes suppressed in carriers of risk allele (as depicted in b); one-tailed hypergeometric test. e, GOBP pathway analysis of genes anti-correlated in patient CD8+ T cells with increasing B cell IL7 expression from the same blood samples; one-tailed hypergeometric test. f, Comparative analysis of z-scores (x axis: B cell IL7 effect, y axis: rs16906115 effect) from pathway analysis in d and e. g, Change in CD8+ T cell mitotic signature score post-sICB according to rs16906115 status; two-sided t-test, n = 65 patients. For box plots in a,c,g, the central line reflects the median value; the box corresponds to the 25–75% quartiles; the upper whisker extends to the largest value no farther than 1.5× IQR; and the lower whisker extends from the 25% quartile to the smallest value no farther than 1.5× IQR.
rs16906115 associates with post-ICB CD8+ T cell clonality
IL-7 is crucial for the survival and proliferation of mature T cell clones28, and we previously observed that clonal CD8+ T cell proportions, both pre-treatment and post-treatment, are prognostic for long-term clinical outcome12,23. To assess whether rs16906115 impacts overall clonal features, we analyzed CD8+ T cell clone sizes, based on unique TCR CDR3 nucleotide sequences, across the cohort (n = 146 CD8+ T cell samples taken immediately before C2, of which 133 had paired baseline samples), blinded to expression phenotype. We first examined the relationship between LS and clone size, and we found that patients with stable LS (increase in count or fall <20%) had significantly more large clones (defined as those comprising >0.5% of the TCR repertoire) both pre-treatment and post-treatment (Fig. 5a), in keeping with both parameters being positively associated with beneficial response to ICB. We subsequently explored the effect of rs16906115 genotype on the Gini index of the TRB chains, a measure of clonal unevenness29, across pre-treatment and post-treatment samples. Notably, we found a significant interaction between ICB treatment and genotype in the change in Gini in response to treatment, with risk allele carriers showing increased clonal inequality post-ICB (Fig. 5b), resultant in skewed clonality after ICB treatment according to allele (Fig. 5c). Finally, we explored the relationship between clonal composition post-treatment according to genotype, and we found that risk allele carriers post-sICB had a repertoire composed of proportionally fewer small clones (<0.05% repertoire in size) and, instead, demonstrated significantly greater repertoire occupancy by large clones (>0.5% repertoire in size) after both cICB and sICB (Fig. 5d).

a, Association between count of large CD8+ T cell clones (defined as those >0.5% repertoire in size using TRB) and LS for same individual pre-ICB (C1, left facet) and immediately before the second cycle of treatment (C2, right facet); two-sided t-test, n = 110 patients (C1), n = 91 patients (C2). b, Gini index (measured on TRB chain) immediately before C2 samples from CD8+ T cells across all ICB treatments with available measurement, stratified by carriage at rs16906115; two-sided t-test, n = 62 sICB patients, n = 76 cICB patients, n = 138 both. c, Change in Gini index between pre-treatment (C1) and post-treatment (C2) by ICB type; two-sided t-test, n = 133. d, Proportion of post-treatment clones per repertoire at C2 defined by TRB CDR3 sequence and V and J gene usage at different size thresholds (left to right facets) and by ICB type (cICB upper, sICB lower), dichotomized by rs16906115 carriage; two-way t-test, n = 77 patients cICB, n = 69 patients sICB. For box plots in a–d, the central line reflects the median; the box corresponds to 25–75% quartiles; the upper whisker extends to the largest value no farther than 1.5× IQR; and the lower whisker extends from the 25% quartile to the smallest value no farther than 1.5× IQR. e, Carriage of rs16906115 minor allele is associated with disease-specific survival in TCGA melanoma dataset; log-rank test. f, Carriage of rs16906115 minor allele is associated with overall survival in TCGA melanoma dataset; log-rank test.
rs16906115 is associated with melanoma survival in TCGA
Given that rs16906115 is associated with baseline pre-treatment differences in patient B cell IL7 expression and B cell maturation, we sought to explore whether this SNP had a relationship with the natural history of melanoma. We examined the frequency of this allele in the TCGA melanoma data series, and we found that it did not differ to that in our cohort (8.3% versus 7.5%, P = 0.72, chi-squared test). We then examined the clinical outcome data from this cohort30, restricting analysis to those whose ethnicity is recorded as ‘white’ and dichotomizing outcomes according to allele carriage (n = 435). Strikingly, we found that carriage of the risk allele was associated with increased progression-free survival (P = 0.016, log-rank test), disease-specific survival (P = 0.015) and overall survival (P = 0.014) (Extended Data Fig. 5a and Fig. 5e,f). This provides further independent evidence as to the importance of this allele in melanoma, inferring that, in addition to predisposing patients to ICB toxicity, B cell IL7 plays a role in the natural history of melanoma.
Discussion
Allelic variation at rs16906115 is a significant risk factor for development of irAEs, most notably in recipients of sICB who have a greatly reduced risk of severe irAEs. Our data replicate the co-reported GWAS by Groha et al. and provide novel insights into mechanisms of response to ICB and irAE risk. The allele rs16906115 is intronic to IL7, and, unexpectedly, we found robust expression of IL7 in B cells, which is markedly induced in patients with melanoma. Notably, patient carriers of the rs16906115 risk allele demonstrate significantly greater induction of B-cell-specific IL7, a context-specific effect not observed in those without melanoma or in other immune subsets31,32,33. Using scRNA-seq, we replicated both patient-specific induction and genotypic association, providing further clarity regarding IL7 expression across B cell subsets and demonstrating altered frequency of these according to rs16906115 status. Notably, supporting the effect of rs16906115 being mediated via B cell IL7, we demonstrate that B cell IL7 is more significantly associated with irAE risk than rs16906115 genotype, with genotypic effect being completely resolved by controlling for B cell IL7. Although we did not observe rs16906115 effects on other circulating immune cells, given that stromal and epithelial cells are key sources of IL-7 (refs. 11,15) and considering the important role of tertiary lymphoid structures in immune responses after ICB34,35, we postulate that other B cell-containing lymphoid tissues may show similar context-specific IL7 expression linked to this allele. We found that rs16906115 is associated with increased LS post-sICB and that LS anti-correlates with induction of naive T cells. Instead, carriage of the risk allele shifts the CD8+ T cell response to ICB from a hyper-proliferative state with expansion of naive subsets to one with reduced overall mitosis and maintenance of differentiated effector subsets, in keeping with known properties of IL-7 and previous observations regarding T cell mitosis and plasma IL-7 (refs. 14,28,36). This is also seen in terms of CD8+ T cell gene pathways differentially expressed in the presence of the risk allele and the regulation of a signature gene set that acts as a proxy for ICB-induced mitosis. Crucially, again in keeping with the effects of rs16906115 on CD8+ T cells being mediated via B cell IL7, we note that the same pathways are repressed by increasing expression of B cell IL7 when correcting for rs16906115, with this association being present in individuals homozygous for the reference allele. Finally, analysis of CD8+ TCR, agnostic to gene expression profiles, corroborates the mechanistic consequences of rs16906115 allelic variation in melanoma, with patients who carry the risk allele displaying distinct clonal responses to treatment, consisting of clonal skewing characterized by fewer small clones but increased repertoire occupancy by expanded large clones. Although IL-7 has been described as an important mediator of naive T cell homeostasis in the resting state, it also has a vital role in T cell activation and memory formation10,25,26,37,38,39. Consequently, these observations suggest the importance of the latter in the context of malignancy-related chronic antigen stimulation and ICB therapy. Although the risk-allele-associated increases in large CD8+ T cell clone populations would be anticipated to provide a more cytotoxic response to both tumor and other tissues, we cannot discount a direct B cell-mediated role for increased irAE risk associated with this allele, supported by increased class switching and immunoglobulin chain mutations in carriers pre-treatment. The large effect size of this polymorphism reflects the central role of IL-7 in both B and T cell lymphopoiesis. Future studies are required to ascertain whether induction of peripheral B cell IL7 is observed in other malignancies, potentially forming a biomarker for immune sensitivity. The finding of improved disease-specific and overall survival in patients with melanoma carrying the risk allele in the TCGA dataset indicates that IL-7 plays a role in the natural history of melanoma and further highlights the potential therapeutic importance of this pathway. This study highlights the power of agnostic genetic analyses to provide insights into human immunity of high relevance to disease and delineates a key role for IL-7 in response to ICB, revitalizing previous proposals for incorporating this molecule as a potential adjunct to immunotherapy strategies36,40,41.
Methods
Participants
Adult patients referred to receive ICB as standard-of-care therapy for the treatment of metastatic melanoma or in the adjuvant setting at Oxford University Hospitals NHS Foundation Trust were prospectively and consecutively approached for recruitment. All patients provided written informed consent to donate samples to the Oxford Radcliffe Biobank (Oxford Centre for Histopathology Research ethical approval reference 19/SC/0173, project nos. 16/A019, 18/A064 and 19/A114) and grant access to their routine clinical data; there was no compensation for this. Patients were recruited between 23 November 2015 and 30 September 2021 and received either cICB (ipilimumab 3 mg kg−1 plus nivolumab 1 mg kg−1 three times a week for ≤4 treatment cycles, followed by maintenance nivolumab) or sICB consisting of nivolumab 480 mg monthly, pembrolizumab 2 mg kg−1 three times a week or pembrolizumab 4 mg kg−1 six times a week. Patient characteristics are given in Extended Data Fig. 1a. Healthy donor participants were recruited via the Oxford Biobank (www.oxfordbiobank.org.uk; ethical approval reference 06/Q1605/55), with written informed consent. All donors were of European ancestry; 104 were female, and 66 were male, aged between 21 years and 66 years (median, 46.5 years; IQR 18 years).
Clinical outcomes
Patient demographic and clinical characteristics were collected from the electronic patient record (EPR). Sex was determined on the self-reported assignment extracted from the EPR. irAEs were reported according to the National Cancer Instituteʼs Common Terminology Criteria for Adverse Events version 4.03. Steroids were defined as a prescription being issued for oral or intravenous corticosteroids to treat irAE(s) according to clinical indication. ‘Prior to cycle 5’ was defined as an irAE occurring before the patient had received a cumulative five doses of ICB and is a metric that has been used previously7. Response data were obtained from the EPR, with progression being defined either clinically or using radiological assessment according to irRECIST.1.1, performed approximately 12 weeks and 24 weeks after initiation of treatment. All patients who had received a minimum of one cycle of ICB were incorporated into the analysis irrespective of clinical outcome.
Sample collection
In total, 30–50 ml of blood was collected into EDTA tubes (BD Vacutainer system) collected immediately before administration of ICB. PBMCs and plasma were immediately obtained from whole blood by density centrifugation (Ficoll Paque). All cell subset isolation for RNA-seq was carried out by magnetic separation (Miltenyi Biotec) using CD8, CD19 and CD14 positive selection for CD8+ T cells, B cells and monocytes and negative selection for natural killer (NK) cells and CD4+ naive cells according to the manufacturer’s instructions, with all steps performed either at 4 °C or on ice. Laboratory blood counts were obtained from the patients’ EPR with pre-treatment samples being defined as those taken closest to the first cycle of ICB, but no more than 30 days beforehand, and post-treatment samples being defined as the first sample taken at least 21 days after ICB, but no later than 49 days. Lymphocyte counts were generated using Sysmex XN series analyzers (Sysmex Corporation) in an NHS clinical diagnostic laboratory.
RNA and DNA extraction
After selection, cells were pelleted and resuspended in 350 μl of RLT Plus buffer with 1% beta-mercaptoethanol or DTT. Samples were snap-frozen at −80 °C for batched RNA extraction. Homogenization of the sample was carried out using a QIAshredder (Qiagen), followed by RNA and gDNA extraction using the AllPrep DNA/RNA/miRNA Kit (Qiagen). RNA and gDNA were eluted into 34 μl or 54 μl of RNase-free water, respectively, and concentration was quantified by Qubit and stored at −80 °C until further analysis.
Genotyping
Genotyping was performed on the Illumina Global Screening Array 24 version 3 (Illumina) and was available from 214 patient participants ≥18 years of age, with sufficient follow-up, who had been diagnosed with metastatic melanoma (n = 203) or started on adjuvant pembrolizumab or nivolumab after recent (<12 weeks) resection of metastatic lymph node or other site disease (n = 11) who had received ≥1 cycles of ICB as a standard-of-care therapy. Genotypes were aligned to b37, filtered for call frequency >0.9, cluster separation >0.3, SNP missingness <3%, MAF >1% and Hardy–Weinberg equilibrium (HWE) P > 10−20, before imputation using the Michigan Imputation Server version 1.2.4 with Haplotype Reference Consortium version 1 as a reference panel, using Eagle2 and Minimac4 for phasing and imputation. After imputation, SNPs were filtered for MAF >4%, HWE P > 10−10 and r2 > 0.7 and lifted-over to b38.
Bulk RNA-seq and analysis
Bulk RNA-seq was performed on CD19+ B cell and CD8+ T cell samples derived from healthy individuals and patients receiving single or combination ICB. RNA was thawed on ice before mRNA isolation using NEBNext Poly(A) mRNA Magnetic Isolation Module Kits. Up to 600 ng of RNA was then used to generate dsDNA libraries using NEBNext Ultra II Directional RNA Library Prep Kits for Illumina (cohort sizes given in the text) as previously described12. Samples were then sequenced on either an Illumina HiSeq 4000 (75-bp paired-end) or a NovaSeq 6000 (150-bp paired-end) at the Oxford Genomics Centre. Differential gene expression and pathway analysis were performed using DESeq2 (ref. 42) version 1.30.1 controlling for batch effects, RNA concentration, age and, where multiple cycles of treatment were incorporated, cycle of treatment. No difference was noted between adjuvant patients and metastatic patients in terms of B cell IL7 expression at baseline (P > 0.2) either pre-treatment or post-treatment. Transcript assembly was performed using StringTie43 using Ensembl GRCh37 as the reference (ftp://ftp.ensembl.org/pub/release-75//gtf/Homo_sapiens.GRCh37.75.gtf.gz) for each sample individually, before merging of assemblies and quantification of isoform expression. Comparative analysis per allele was based on read counts using a Wilcoxon signed-rank test. Mitotic signature score was calculated using DESeq2-normalized expression data, taking the geometric mean of the top 50 genes most associated with the mitotic subset in previous CD8+ T cell single-cell sequencing data23.
Adaptive receptor analysis
B cell receptor (BCR) and TCR analysis was performed using the MiXCR44 version 3.0.13 package with settings as previously described12,23. Clonal metrics were calculated from TCR and BCR data by taking the minimum number of reads/chains recorded across samples and re-sampling other samples to the same depth, bootstrapping 1,000 times. BCR usage was generated for IGHC genes. Usage was calculated as either the percentage of IGH clones or reads expressing each individual IGHC gene segment. Mutation rate was inferred in BCR samples, representative of junctional diversity and somatic hypermutation. Information regarding base substitutions, insertions and deletions was extracted from MiXCR output files from ‘allVAlignments’, ‘allDAlignments’ and ‘allJAlignments’ columns. The number of mutations per BCR clone was calculated as the number of substitutions, insertions and deletions combined. Average mutation percentage was then calculated per BCR sample as the total number of mutations detected per clone divided by the total BCR length. Analysis was repeated by chain.
scRNA-seq
5′ scRNA and VDJ sequencing (10x Genomics) was performed on PBMCs (kit version 2) according to the manufacturer’s instructions. In brief, cryopreserved PBMCs were thawed and purified using the Dead Cell Removal Kit. Cells from 5–8 donors were pooled, and an estimated 50,000–60,000 cells were loaded per channel. Gene expression and VDJ libraries were 150-bp paired-end sequenced on a NovaSeq 6000 (Illumina) with a targeted read depth of 25,000 reads per cell. Reads were mapped using cellranger version 6.0.1, with the union of cells called by cellranger and EmptyDroplets45 used downstream. Donor identification was performed using cellsnp-lite and vireoSNP46, and ambient RNA contamination was removed with SoupX47. Cells identified as genotypic doublets, and those with nFeatures <300 or mitochondrial reads >20%, were excluded. The remaining PBMCs were separately clustered and projected into two-dimensional (2D) uniform manifold approximation and projection (UMAP) space in Seurat version 4.1 (ref. 48), using Harmony49 for pool harmonization. After quality control (including removal of doublets and exclusion of TCR or dual-BCR carrying cells), clusters identified as B cells based on marker expression (that is, CD79A and MS4A1) and subset inference using SingleR50 were retained for further analysis. Subset clusters were annotated based on a combination of gene expression, SingleR inferences and VDJ usage, and IL7 expression was imputed using MAGIC51.
Flow cytometry
Patient PBMCs frozen in 90% FCS + 10% DMSO were thawed at 37 °C and washed once in HBSS before staining of 1 × 106 cells with either LIVE/DEAD Fixable Near-IR (Invitrogen) or Zombie Green (BioLegend). Cells were washed and subsequently stained with antibodies against CD3, CD4, CD8α, CD27 and CD45RA in 5% FBS + HBSS. Alternatively, cells were stained with antibodies targeted against CD19, CD3, CD56 and CD14 along with biotinylated anti-IL-7 before staining with streptavidin-PE. Fluorescence minus one (FMO) controls along with primary biotinylated anti-IL-7 and secondary streptavidin-PE alone controls were included to determine IL-7 gating per individual. After washing, cells were fixed for 10 minutes with 2% paraformaldehyde (Sigma-Aldrich), washed and resuspended in 5% FBS + HBSS before acquisition on a BD LSRII. Data were analyzed in FlowJo (Tree Star, version 10.7.1) and R (version 4.0.5). Antibody details are shown in Supplementary Table 9, and gating was as previously performed12. All staining steps were performed for 30 minutes and at 4 °C unless otherwise stated.
General statistical analysis
Statistical comparisons were performed as indicated in each figure legend and conducted in R (version 4.0.5), with graphics using ggplot2 version 3.3.5. Paired tests were used for paired samples, usually using Wilcoxon signed-rank test as indicated. For unpaired samples, the Wilcoxon rank-sum test was primarily used as per the figure legends. ORs were generated using a generalized linear model (family = ‘binomial’). Random effect mixed linear models were run in lme4 version 1.1-26 using lmerTest version 3.1-3 to explore association between B cell IL7 and irAE development, controlling for rs16906115 and ICB type. Survival analysis was performed using survival version 3.2-11. Pathway analysis was performed using XGR version 1.1.8. Adjusted P values are reported using Benjamini–Hochberg correction for multiple comparisons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Cell-specific expression data are made freely available via EGA (EGAC00001001482) upon completion of a data access agreement that can be obtained by contacting the corresponding author.
Code availability
All scripts used to create figures, including the minimal dataset required to recreate figures, consisting of gene expression count data matrices, flow cytometry counts, genotyping at rs16906115 and scRNA-seq data of B cells in the form of a Seurat object, are available via the Fairfax Group bitbucket (https://bitbucket.org/Fairfaxlab/).
References
Larkin, J. et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 381, 1535–1546 (2019).
Weber, J. et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 377, 1824–1835 (2017).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 20, 1239–1251 (2019).
Michot, J. M. et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54, 139–148 (2016).
Schneider, B. J. et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J. Clin. Oncol. 39, 4073–4126 (2021).
Ramos-Casals, M. et al. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 6, 38 (2020).
Ye, W. et al. Checkpoint-blocker-induced autoimmunity is associated with favourable outcome in metastatic melanoma and distinct T-cell expression profiles. Br. J. Cancer 124, 1661–1669 (2021).
Widmer, M. B. et al. Lymphopoiesis and IL‐7. Int. J. Cell Cloning 8, 168–172 (1990).
Clark, M. R., Mandal, M., Ochiai, K. & Singh, H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 14, 69–80 (2014).
Fry, T. J. & Mackall, C. L. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J. Immunol. 174, 6571–6576 (2005).
Martin, C. E. et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity 47, 171–182 (2017).
Fairfax, B. P. et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med. 26, 193–199 (2020).
Huang, A. C. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017).
Shive, C. L. et al. Inflammation perturbs the IL-7 axis, promoting senescence and exhaustion that broadly characterize immune failure in treated HIV infection. J. Acquir. Immune Defic. Syndr. 71, 483–492 (2016).
Miller, C. N. et al. IL-7 production in murine lymphatic endothelial cells and induction in the setting of peripherallymphopenia. Int. Immunol. 25, 471–483 (2013).
Tissue expression of IL7. Summary. The Human Protein Atlas. https://www.proteinatlas.org/ENSG00000104432-IL7/tissue
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Bates, D., Mächler, M., Bolker, B. M. & Walker, S. C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Morgan, B. et al. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiatisn. EMBO J. 16, 2004–2013 (1997).
Hodson, D. J. et al. Regulation of normal B-cell differentiation and malignant B-cell survival by OCT2. Proc. Natl Acad. Sci. USA 113, E2039–E2046 (2016).
Luo, Y., Fujii, H., Gerster, T. & Roeder, R. G. A novel B cell-derived coactivator potentiates the activation of immunoglobulin promoters by octamer-binding transcription factors. Cell 71, 231–241 (1992).
Fang, H., Knezevic, B., Burnham, K. L. & Knight, J. C. XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med. 8, 129 (2016).
Watson, R. A. et al. Immune checkpoint blockade sensitivity and progression-free survival associates with baseline CD8+ T cell clone size and cytotoxicity. Sci. Immunol. 6, eabj8825 (2021).
Subudhi, S. K. et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc. Natl Acad. Sci. USA 113, 11919–11924 (2016).
Gagnon, J. et al. Increased antigen responsiveness of naive CD8 T cells exposed to IL‐7 and IL‐21 is associated with decreased CD5 expression. Immunol. Cell Biol. 88, 451–460 (2010).
Schluns, K. S., Kieper, W. C., Jameson, S. C. & Lefrançois, L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 1, 426–432 (2000).
Rutishauser, R. L. et al. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31, 296–308 (2009).
Carrette, F. & Surh, C. D. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin. Immunol. 24, 209–217 (2012).
Chiffelle, J. et al. T-cell repertoire analysis and metrics of diversity and clonality. Curr. Opin. Biotechnol. 65, 284–295 (2020).
Liu, J. et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416 (2018).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014).
Kim, S. et al. Characterizing the genetic basis of innate immune response in TLR4-activated human monocytes. Nat. Commun. 5, 5236 (2014).
Lee, M. N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Lynch, D. H. & Miller, R. E. Interleukin 7 promotes long-term in vitro growth of antitumor cytotoxic T lymphocytes with immunotherapeutic efficacy in vivo. J. Exp. Med. 179, 31–42 (1994).
Gagnon, J. et al. IL-6, in synergy with IL-7 or IL-15, stimulates TCR-independent proliferation and functional differentiation of CD8+ T lymphocytes. J. Immunol. 180, 7958–7968 (2008).
Valbon, S. F., Condotta, S. A. & Richer, M. J. Regulation of effector and memory CD8+ T cell function by inflammatory cytokines. Cytokine 82, 16–23 (2016).
Rathmell, J. C., Farkash, E. A., Gao, W. & Thompson, C. B. IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 167, 6869–6876 (2001).
Pellegrini, M. et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat. Med. 15, 528–536 (2009).
ElKassar, N. & Gress, R. E. An overview of IL-7 biology and its use in immunotherapy. J. Immunotoxicol. 7, 1–7 (2010).
Love, M. I., Anders, S. & Huber, W. Differential analysis of count data—the DESeq2 package. https://bioc.ism.ac.jp/packages/3.4/bioc/vignettes/DESeq2/inst/doc/DESeq2.pdf (2016).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Bolotin, D. A. et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381 (2015).
Lun, A. T. L. et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63 (2019).
Huang, Y., McCarthy, D. J. & Stegle, O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019).
Young, M. D. & Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9, giaa151 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Acknowledgements
We are very grateful to all patients who contributed samples and participated in the study. We thank all the staff of the Day Treatment Unit, Oxford Cancer Centre and the Brodey Centre at the Horton General Hospital. We are grateful to all the staff of the Oxford University Hospitals NHS Foundation Trust haematology and biochemistry laboratory and thank T. James for his facilitation, as well as the staff of the Oxford Radcliffe Biobank and Churchill Hospital Sample Handling Lab. This study was funded by a Wellcome Intermediate Clinical Fellowship to B.P.F. (no. 201488/Z/16/Z), additionally supporting A.V.A. and I.N. R.A.W. is funded by a Wellcome Trust Doctoral Training Fellowship (no. BST00070). O.T. is supported by the Clarendon Fund, St. Edmund Hall and an Oxford Australia Scholarship. W.Y. is an NIHR academic clinical fellow and is supported by a CRUK predoctoral fellowship (reference RCCTI\100019). R.C. was supported by a CRUK Clinical Research Training Fellowship (S_3578). C.A.T. was funded by the Engineering & Physical Sciences Research Council and the Balliol Jowett Society (no. D4T00070). D.C. is funded by a Cancer Research UK Advanced Clinician Scientist Fellowship (C26642/A27963). J.J.G. is supported by an NIHR Clinical Lectureship. M.R.M. and B.P.F. are supported by the NIHR Oxford Biomedical Research Centre. K.M. and P.E. are supported by the NIHR Leeds Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
B.P.F., V.N. and A.G. conceived the study. N.C., M.J.P., M.R.M., B.P.F., V.W. and R.A.W. recruited patient participants. B.P.F., S.K., C.A.T., R.A.W., K.M. and P.E. recruited healthy volunteer participants. C.A.T., R.A.W., O.T., R.C., W.Y., A.V.A., P.K.S., S.K., R.C. and E.J. collected and processed blood samples. C.A.T., O.T., A.V.A. and W.Y. performed genotyping. C.A.T. and A.V.A. generated RNA-seq libraries. R.A.W., W.Y. and B.P.F. collected clinical follow-up data. B.S. collected laboratory blood results. C.A.T. performed flow cytometry experiments. D.C. assisted with TCGA analysis. O.T., W.Y. and A.V.A. performed single-cell sequencing. C.A.T., R.A.W., O.T., W.Y., J.J.G. and B.P.F. performed data analysis. B.P.F. drafted the manuscript, with support from R.A.W. V.N., M.R.M., T.C. M.F. and A.G. contributed to discussions throughout the work. B.P.F. oversaw the project. All authors read, edited and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
C.A.T., R.A.W., O.T., W.Y., R.C., A.V.A., P.K.S., I.N., J.J.G. and A.G. declare no competing interests. M.R.M. reports grants from Roche, grants from AstraZeneca, grants and personal fees from GSK, personal fees and other from Novartis, other from Millenium, personal fees and other from Immunocore, personal fees and other from BMS, personal fees and other from Eisai, other from Pfizer, personal fees, non-financial support and other from Merck/MSD, personal fees and other from Rigontec (acquired by MSD), other from Regeneron, personal fees and other from BiolineRx, personal fees and other from Array Biopharma (now Pfizer), non-financial support and other from Replimune, personal fees from Kineta and personal fees from Silicon Therapeutics, all outside the submitted work. D.C. reports funding from Haliodx and consultancy for Merck. K.S. reports grants from Gilead, Eli Lilly and BMS and personal fees from UCB, AZD, Abbvie and Eli Lilly. P.E. performed consultancy for Abbvie, AstraZeneca, BMS, Boehringer Ingelheim, Galapagos, Gilead, Janssen, MSD, Eli Lilly, Novartis, Pfizer, Roche and Samsung and was involved in clinical trials for Abbvie, BMS, Eli Lilly, Novartis, Pfizer, Roche and Samsung. T.K.C. reports research/advisory boards/consultancy/honorarium (institutional and personal, paid and unpaid) from AstraZeneca, Aveo, Bayer, BMS, Eisai, EMD Serono, Exelixis, GSK, IQVA, Ipsen, Kanaph, Eli Lilly, Merck, Nikang, Novartis, Pfizer, Roche, Sanofi/Aventis, Takeda and Tempest. Travel, accommodations, expenses, medical writing in relation to consulting, advisory roles or honoraria. Stock options: Pionyr and Tempest. Other: up-to-date royalties, CME-related events (for example, OncLIve, PVI and MJH Life Sciences) honoraria. NCI GU Steering Committee. Patents filed, royalties or other intellectual properties (no income as of current date): related to biomarkers of immune checkpoint blockers and ctDNA. No speaker’s bureau. B.P.F. received conference support from BMS and performed consultancy for UCB.
Peer review
Peer review information
Nature Medicine thanks Zlatko Trajanoski, Leng Han and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary handling editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Extended Clinical Data.
(a) Details of patient cohort, tests are as specified and were two-sided (b) Comparative response of lymphocyte count to cICB and sICB; two-sided Wilcoxon signed rank test, n = 134 patients (74 cICB, 60 sICB). c) Comparison of fall in lymphocyte count in recipients of nivolumab versus those of pembrolizumab; two-sided T tests (n = 74 ipi/nivo, n = 9 nivo, n = 51 pembrolizumab). (d) Kaplan Meier analysis of progression-free survival in immunotherapy naïve metastatic melanoma patients within the cohort according to LS (unstable defined as post-treatment lymphocyte count ≤80% pre-treatment value); log-rank test (n = 111). (e) As per (d), but curves represent overall survival across cohort according to LS (n = 111). For boxplots in Extended Data Fig. 1b, c central line reflects the median, the box reflects the 25%–75% quartiles, upper whisker extends to largest value no further than 1.5x interquartile range, lower whisker extends from 25% quartile to the smallest value no further than 1.5x interquartile range.
Extended Data Fig. 2 IL7 Expression Across Cell Types.
(a) Association between donor age and IL7 expression across cell types, stratified by status (HD – healthy donor); P value from two-sided ANOVA, correcting for status (CD14: n = 42 HD, 216 patient samples; CD19: n = 90 HD, 159 patient samples; CD4 naïve: n = 8 HD, 50 patient samples; CD8: n = 68 HD, 326 patient samples; NK: n = 31 HD, 120 patient samples). (b) B cell expression of IL7 in patient samples according to cycle and treatment type, no significant difference was noted with type or between baseline untreated and immediately prior to the second cycle of treatment (C2); two-sided T test, n = 113 cICB, n = 46 sICB. (c) B cell expression of IL7 split by treatment type according to rs16906115 carriage; one-sided Wilcoxon rank-sum test, C1: n = 64 cICB patients, n = 27 sICB patients; C2: n = 49 cICB patients, n = 19 sICB patients; late: n = 24 cICB patients, n = 15 sICB patients. (d) Expression of IL7 in patient and healthy donor (HD) samples dichotomized by rs16906115 status, faceted by cell type, across all samples in cohort, P value refers to result from two-sided T test between absence and presence of risk allele (one or two copies). (e) Expression of IL7 transcripts annotated using StringTie43 according to allele status across all B cell samples showing a pan transcript effect of rs16906115 carriage; two-sided Wilcoxon rank-sum test. (f) Representative flow plot showing PBMCs gated on expression of CD19 with forward scatter on the X-axis versus IL-7 positive events on the y-axis. Primary = primary biotinylated anti-IL-7, secondary = Streptavidin PE, FMO = fluorescence minus one. (g) Analysis of samples demonstrated consistent detection of IL-7 positive B cell population in PBMCs from patients (n = 2) and controls (n = 2), with IL-7 positive fractions not detected in cells gated for CD3 (T cells), CD56+, CD3- (NK cells) or CD14 (monocytes). For boxplots in Extended Data Fig. 2b–e, g; the central line reflects the median, the box 25%–75% quartiles, upper whisker extends to largest value no further than 1.5x interquartile range, lower whisker extends from 25% quartile to the smallest value no further than 1.5x interquartile range.
Extended Data Fig. 3 B cell correlates of IL7 expression.
(a) GOBP pathway analysis of genes anti-correlated with B cell IL7 (FDR < 0.05), x -axis demonstrates fold-change above background, y-axis z score, all points above dotted line FDR < 0.01, one-tailed hypergeometric test. (b) Percentage chains detected with mutation from germline (chain denoted in label) or clones containing IGHD (y axis) versus relative expression IL7 for that sample (x axis), P value refers to Spearman rank-test result, dotted lines reflect fitted linear model, shaded area 95% Confidence Interval. (c) Heatmap of key B cell subset specific genes from scRNA-seq data. (d) GOBP analysis of genes differentially expressed between B cell subsets, top 5 pathways per subset listed, one-tailed hypergeometric test. (e) UMAP from main Fig. 3e, depicting relative IL7 expression per cell. (f) Relative expression of IL7 (imputed) using scRNA-seq dataset across all B cells separated by subset (n = 28 samples). (g) Immunoglobulin chain usage in Naïve B cell subset according to patient status (HD - healthy donor n = 5, mel – metastatic melanoma n = 23); two-sided Wilcoxon rank-sum test. (h) Median expression of IL7 per B cell subset from scRNA-seq data dichotomised by rs16906115 demonstrating genotype acts through altering proportions of subsets as opposed to relative expression per cell (n = 23 patients); two-sided Wilcoxon rank-sum test. For boxplots in Extended Data Fig. 3f–h the central line reflects the median value, the box corresponds to 25%–75% quartiles, the upper whisker extends to largest value no further than 1.5x interquartile range, the lower whisker extends from 25% quartile to the smallest value no further than 1.5x interquartile range.
Extended Data Fig. 4 Extended T cell data.
(a) Correlation of fold change in lymphocyte count measured on routine hospital bloods (as per Lymphocyte Stability) versus % change in cell subset as measured using flow-cytometry at the same timepoint. From left: right; % all T cells in PBMC, % T Effector Memory, % Naïve T cells; P value from Spearman rank test, dotted lines reflect fitted linear model, shaded area the 95% Confidence Interval, n = 40. (b) Correlation of CD8+ T cell expression of SH3BGRL2 versus the naïve marker CD27 in healthy donors (left facet) and patient samples (right facet); Spearman rank test, shaded area reflects 95% Confidence Interval of fitted linear model, P interaction with status <0.001, n = 67 controls, n = 326 patient C1/C2 samples. (c) GOBP pathway analysis of genes upregulated in CD8+ T cells in carriers of risk allele (as depicted in Fig. 4b), one-tailed hypergeometric test. (d) GOBP pathway analysis of genes anti-correlated in patient CD8+ T cells with increasing B cell IL7 expression from same blood samples, having corrected for rs16906115 status, one-tailed hypergeometric test. e) GOBP pathway analysis of genes anti-correlated in patient CD8+ T cells with increasing B cell IL7 expression in non-carriers of risk allele only, one-tailed hypergeometric test.
Extended Data Fig. 5 TCGA survival data by rs16906115 carriage.
(a) Progression-free survival according to rs16906115 allele status across All Stages in TCGA melanoma dataset. (b) As per Extended Data Fig. 5a, but Stages IIB-IV. (c) Carriage of rs16906115 minor allele is associated with disease-specific survival in Stages IIB-IV in TCGA melanoma dataset. (d) Overall Survival according to rs16906115 allele status across Stages IIB-IV Stages in TCGA melanoma dataset. All P values correspond to log-rank test.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2
Supplementary Table
Supplementary Tables 1–9
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taylor, C.A., Watson, R.A., Tong, O. et al. IL7 genetic variation and toxicity to immune checkpoint blockade in patients with melanoma. Nat Med 28, 2592–2600 (2022). https://doi.org/10.1038/s41591-022-02095-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-022-02095-5
This article is cited by
-
Clinical and translational attributes of immune-related adverse events
Nature Cancer (2024)
-
Drug-microbiota interactions: an emerging priority for precision medicine
Signal Transduction and Targeted Therapy (2023)
-
Using genetics to predict toxicity of cancer immunotherapy
Nature Medicine (2022)
