
Abstract
Idiopathic pulmonary fibrosis (IPF) is a lethal fibrosing interstitial lung disease with a mean survival time of less than 5 years. Nonspecific presentation, a lack of effective early screening tools, unclear pathobiology of early-stage IPF and the need for invasive and expensive procedures for diagnostic confirmation hinder early diagnosis. In this study, we introduce a new screening tool for IPF in primary care settings that requires no new laboratory tests and does not require recognition of early symptoms. Using subtle comorbidity signatures identified from the history of medical encounters of individuals, we developed an algorithm, called the zero-burden comorbidity risk score for IPF (ZCoR-IPF), to predict the future risk of an IPF diagnosis. ZCoR-IPF was trained on a national insurance claims database and validated on three independent databases, comprising a total of 2,983,215 participants, with 54,247 positive cases. The algorithm achieved positive likelihood ratios greater than 30 at a specificity of 0.99 across different cohorts, for both sexes, and for participants with different risk states and history of confounding diseases. The area under the receiver-operating characteristic curve for ZCoR-IPF in predicting IPF exceeded 0.88 and was approximately 0.84 at 1 and 4 years before a conventional diagnosis, respectively. Thus, if adopted, ZCoR-IPF can potentially enable earlier diagnosis of IPF and improve outcomes of disease-modifying therapies and other interventions.
Similar content being viewed by others
Main
IPF is an irreversible, progressive, debilitating and ultimately lethal fibrosing interstitial lung disease (ILD) of unknown cause1,2,3. Before the introduction of the anti-fibrotic medications nintedanib and pirfenidone in 2014, the typical survival of individuals with IPF was 2–5 years from the time of diagnosis4. This prognosis has been characterized as worse than that of most cancers5. Although the disease is considered rare, as of 2014, IPF had a greater worldwide prevalence than did all but the seven most common cancers5. Over the past decade, timely, efficient and confident diagnosis of IPF has been recognized as a major public health challenge worldwide2,5,6,7,8,9,10,11,12,13,14. In this study, we develop a screening tool to predict future IPF diagnosis by learning subtle patterns in the time, nature and ordering of past medical encounters of individuals with IPF.
Identifying IPF cases is a complex, multistep and often multiyear process9,11,12,13,14,15,16. A usually necessary but not always sufficient condition is referral for high-resolution computed tomography (HRCT) of the chest, often at an expert center9,13. In most cases, such referral leads to eventual recognition of radiologic or histologic usual interstitial pneumonia (UIP), the hallmark of IPF, or of other radiologic or histologic signs associated with this disease, and serologic studies to rule out other forms of ILD2,9,13. For such referral to happen, one or more of a number of scenarios must unfold: (1) participants recognize the chronicity, progression or both of respiratory symptoms and the resultant need for medical attention13,14,16,17; (2) health-care practitioners note the importance of these symptoms, of signs of possible fibrosis on auscultation or of both5,18; (3) radiologists, not infrequently non-specialists in chest imaging, note incidental findings of interstitial lung abnormalities (ILAs) or ILD on thoracic or abdominal CT8,12,19,20. Alternatively, a pulmonologist or a HRCT referral may only take place20 after an emergency room visit or an acute exacerbation of IPF14. Hence, IPF diagnosis is often delayed by multiple physician visits and repeated, sometimes invasive tests11, and misdiagnosis rates approaching 40% are reported13.
Reliable early diagnosis of IPF is hindered by nonspecific clinical symptoms16,17, for example, insidiously progressive, chronic, exertional dyspnea and/or chronic, often mild cough. These symptoms are easily attributed by individuals to age or deconditoning13, or by physicians to more common cardiorespiratory diseases5,13,14,15. Important risk factors for IPF, namely, older age, male sex and cigarette smoking1, are similarly nonspecific. Notably, the current diagnostic hallmark of IPF, UIP on HRCT or histology2, is a late-stage finding3, and our limited understanding and characterization of phenotypic and genetic findings associated with ‘early-stage IPF’8 also hinder early diagnosis. Moreover, UIP may be confirmed via relatively invasive procedures requiring specialized interpretation, for example, HRCT or surgical lung biopsy13. Finally, no validated or easily applicable screening modalities currently exist for IPF7.
In this study, we introduce ZCoR-IPF as a screening tool with the potential to ameliorate these key challenges. This tool requires no new diagnostic tests, may be universally administered and does not necessarily require recognition of early symptoms by the participants or care providers. Analyzing large databases of electronic health records (EHRs) via new pattern discovery algorithms, we identify subtle comorbidity incidence, timing and sequence characteristics presaging IPF. Combining these discovered features with state-of-the-art machine learning then leads to a powerful, accurate, automated screening tool based only on diagnostic codes, age and sex that exist already in the participant’s past medical record. Here, we report on the training of ZCoR-IPF on a large national insurance claims database, and validation on held-back data and on de-identified records from two additional datasets. Our results indicate that ZCoR-IPF can accurately detect IPF risk in individuals with data available at the point of care and thus has the potential to be deployed within primary care workflows for universal at-scale IPF screening.
Results
Data source and participant selection
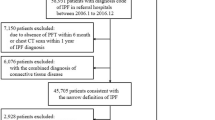
We considered participants from three different databases. The first dataset (referred to as the Truven dataset) is a part of the Truven Health Analytics MarketScan Commercial Claims and Encounters Database for 2003–2018 (ref. 21). This US national database merges data contributed by over 150 insurance carriers and large self-insurance companies and comprises over 7 billion time-stamped diagnosis codes. About 6.6% of participants are covered by Medicare. The Truven database tracks over 87 million participants for periods ranging from 1 to 15 years, reflecting a substantial cross-section of the US population. We selected the cohort(s) in accordance with the inclusion and exclusion criteria described in Table 1, ensuring that selected participants had at least 3 years of medical history recorded in the database. The geographical distribution of the participants in the selected cohort(s) is illustrated in Fig. 1a, which correlates with state-specific population density in the United States. Figure 1b shows the age distribution at the time of IPF diagnosis (mean ≈ 68 years), which is consistent with the reported mean onset age for IPF (66 years4). We also note that observed risk of onset increases with age, which is computed as the number of IPF cases normalized by the total number of participants at the same age (Fig. 1c). Figure 1d shows the participant selection via a CONSORT (Consolidated Standards of Reporting Trials) diagram22 for the Truven dataset.

a, Geographical origin within the United States of male and female participants in the Truven dataset. b, Distribution of participant ages at the time of diagnosis (onset age) in the database. c, The probability of diagnosis with age, taking into account the variation of the number of participants of a given age in the database (the higher the age beyond 65 years, the smaller the number of participants). d,e, Participant selection in the Truven (d) and UCM (e) databases shown using CONSORT diagrams.
While the Truven dataset is used for both training and out-of-sample cross-validation with held-back data, the second independent dataset comprises de-identified diagnostic records of participants treated at the University of Chicago Medical Center between 2006 and 2021 (referred to as the UCM dataset) and aids in independent cross-validation. We considered participants aged 45 years and older and applied the same inclusion/exclusion criteria as the Truven dataset. Figure 1e is the corresponding CONSORT diagram for the UCM dataset, showing that 68,658 UCM participants were analyzed.
The third dataset (referred to as the MAYO dataset) comprises a random sample drawn from the de-identified administrative claims data from the OptumLabs Data Warehouse23, which includes medical and pharmacy claims, and enrollment records for commercial and Medicare Advantage enrollees. The database contains longitudinal health information on participants collected between the years 2010 and 2020, representing a diverse mixture of ages, ethnicities and geographical regions across the United States, and has almost equal representation across males and females. Among Medicare Advantage enrollees, females are somewhat overrepresented (58% females versus 42% males). Geographically, a higher percentage of enrollees were from the South (48%) compared to 25% from the Midwest, 9% from the West and 18% from the Northeast. Unlike the first two sources for which the whole team had access to complete de-identified participant records and characteristics, analysis of the MAYO data was designed to simulate a pragmatic clinical workflow. The compiled code for ZCoR-IPF was shared with Mayo Clinic personnel, who applied the tool on a large random selection from the OptumLabs Data Warehouse (aged between 45 and 90 years), and reported back the performance metrics, eliminating any direct contact between raw participant data and ZCoR-IPF developers, and hence the possibility of dataset-specific selection and other biases. The MAYO results were reported only for participants who passed the inclusion/exclusion criteria described above. We analyzed a total of 861,280 participants from the MAYO dataset, of whom 315 had an IPF diagnosis.
Thus, in total, this study considers n = 2,983,215 participants (Truven: 2,053,277; UCM: 68,658; MAYO: 861,280), with npositive = 54,247 IPF diagnoses. We did not enumerate participant characteristics in the MAYO dataset, where third-party processing precluded collating detailed participant information.
Electronic health record data processing
We viewed the task of predicting a future IPF diagnosis as a binary classification problem, and began by segregating time-stamped sequences of diagnostic codes into positive and control categories, where the ‘positive’ category refers to participants diagnosed with IPF 1 year (Extended Data 1) from the point of screening. We also considered earlier screening up to 4 years before the clinical diagnosis. The control cohort comprised participants lacking have any target codes in their records, within the 2 years after the point of screening. For both groups, we based our predictions on the past 2 years of medical history.
The International Classification of Diseases (ICD) diagnostic codes specifically for IPF are 516.31 (Ninth Revision (ICD-9)) and J84.112 (Tenth Revision (ICD-10)). Here, we primarily identified IPF participants as those with either one of these two ICD codes in their medical record (Extended Data 1; ‘narrow target’), aiming to predict an IPF diagnosis via elevated values of the ZCoR-IPF score, before one of these codes showed up in the medical history. To account for coding uncertainties in administrative data, we also considered (1) a secondary analysis with a broader target definition encompassing ILD and related disorders (Extended Data 1; ‘broad target’), and (2) restricted positive cohorts where confidence in IPF diagnosis is increased by looking for either IPF-specific prescriptions (pirfenidone or nintedanib; the IPF-Rx sub-cohort) or signatures of associated codes that typically indicate pathways to IPF diagnosis (the IPF-Ax sub-cohort; Methods). ZCoR-IPF achieved comparable performance across all these cases.
Importantly, we did not preselect or reject any diagnostic or prescription code based on its known or suspected comorbidity with IPF. To investigate if our predictive performance changed substantially for participants who are at ‘high risk’ based on known comorbidities, we also evaluated our performance within a high-risk and a low-risk sub-cohort. The high-risk sub-cohort comprised participants with one or more of the diagnoses enumerated in Extended Data 2, which identify the top-known IPF comorbidities24. Our results for the low-risk sub-cohort (defined as participants not at ‘high risk’) is of particular significance, as IPF-positive participants in this cohort are more likely to be missed. Additionally, we investigated ZCoR-IPF applicability in participants (1) for whom IPF diagnosis might be confounded due a preexisting condition such as chronic obstructive pulmonary disease (COPD), a heart condition or asthma (Supplementary Table 1 for cohort definition codes) and (2) for whom the absence of any indication of dyspnea presumably reduces the odds of IPF suspicion delaying diagnosis (Supplementary Table 2).
ZCoR-IPF modeling, training and prediction
We partitioned the disease spectrum into 51 broad categories, for example, infectious diseases, immunologic disorders and endocrine disorders (Supplementary Table 3), approximately aligning with the categories defined within the ICD framework25. Each of the diagnostic categories yields a single time series over weeks (each week was identified as having a value ’0’ for no code corresponding to the diagnostic category, or ’1’ if some code from that category was present, and ’2’ if a diagnostic code from any of the other categories was present). We refer to the individual diagnostic categories as a phenotype, because they are observable characteristics of the participants. With these diagnostic phenotypes, each participant is represented by 51 sparse stochastic time series of events, which are compressed into specialized hidden Markov models (HMMs) known as probabilistic finite automata26 (PFSA). These models are inferred separately for each phenotype, for each sex and for the control and the positive cohorts, to identify the distinctive average patterns emerging at the population level. Thus, we inferred 51 × 2 × 2 = 204 PFSA models in total in this study (Methods). Importantly, PFSA models are ideal for capturing complex stochastic and long-range longitudinal effects of past medical encounters.
Given these models, and a specific participant, we can quantify the likelihood of this participant’s particular history being generated by the control PFSA models as opposed to the positive models. We refer to this likelihood difference as the sequence likelihood defect (SLD)27, which is a key informative feature in our approach. Ultimately, we computed 667 features (Extended Data 3) for each participant (all of these features are functions of data available at the point of care), which inform the ZCoR-IPF score. We randomly chose 75% of the participants in the Truven dataset for training with the rest held out as a validation set. Random splitting of the dataset resulted in nearly identical participant characteristics in training and test data (Table 2).
Performance measurement
We measured our performance using the area under the receiver-operating characteristic (ROC) curve (AUC), sensitivity, specificity, the positive and negative predictive values (PPV and NPV, respectively) and the positive and negative likelihood ratios (LR+ and LR−, respectively). The PPV and NPV depend on the prevalence of the disease, while the likelihood ratios do not. Because IPF is relatively rare, achieving a high PPV is difficult28. Indeed, likelihood ratios are more useful in quantifying the effectiveness of a new screening tool: LR+ is the ratio of the likelihood of the disease given a positive ZCoR-IPF flag, to the likelihood of the disease before ZCoR-IPF screening, and similarly, LR− is the ratio of the likelihood of a participant not developing the target disorder given a negative ZCoR-IPF result, to the likelihood of the disease before ZCoR-IPF screening. An effective screening tool has a large LR+ value (>10.0 is deemed excellent29,30), and a small LR− value (<1.0).
ZCoR-IPF performance
We demonstrate that ZCoR-IPF has robust out-of-sample performance, allowing (1) accurate out-of-sample predictions for a future IPF diagnosis via leveraging subtle comorbidity patterns recorded in individual histories (Fig. 2, Tables 3 and 4 and Extended Data 4 and 5), (2) maintenance of high predictive performance for diagnosis further out, up to 4 years in future, and (3) common confounders to have little or no effect, such as a preexisting diagnosis of COPD, asthma or heart disease or the absence of any indication of dyspnea in the past. Additionally, we show that ZCoR-IPF outperforms baseline predictors and state-of-the-art neural network (NN) or deep learning architectures trained in the same manner as ZCoR-IPF (Methods). Results for the baseline model (a logistic regressor with 87 suspected IPF risk factors listed in Supplementary Table 4) on the Truven and the UCM datasets (Extended Data Fig. 1) demonstrate that ZCoR-IPF is significantly (nonoverlapping 95% confidence interval (CI)) and substantially better. Extensive comparison with state-of-the-art NN models demonstrated either poor out-of-sample performance or performance that fails to replicate across databases (Extended Data Fig. 2 and Supplementary Table 5), although some specific code embedding or a particular choice of NN architecture might mitigate these issues. We also found that substantially delayed updates to participant records, for example, no data available on medical encounters for up to the past 2 months before the point of screening, do not impact ZCoR-IPF performance (Extended Data Fig. 1 and Supplementary Table 6), establishing its applicability in the primary care workflow.

a, Out-of-sample ROC curves. b, Out-of-sample precision–recall curves. For out-of-sample participants from the Truven dataset, we achieved AUCs > 88% for both sexes, with sensitivity > 52% (males) and > 60% (females) at 95% specificity. The PPV values obtained were ≈ 73% (males) and 67% (females) at 99.7% specificity assuming a uniform prevalence of 5%. c, The variation of positive likelihood ratio (LR+) with the negative likelihood ratio (LR−), illustrating LR+ > 30 with a low LR− (≦ 0.7) for all sub-cohorts. Analyses in a and c are independent of population prevalence of IPF. d, Variation of PPV with NPV, assuming a uniform prevalence of 5%. e, Top 20 comorbidity categories sorted in the order of inferred importance.
Key results for the primary analysis are presented in Fig. 2a,b, illustrating the ROC and the precision–recall curves (for screening 1 year before current diagnosis), shown separately for males and females. Tables 3 and 4 and Extended Data 4 and 5 enumerate the performance metrics computed for different subsets, for male and female cohorts, evaluated at 95% and 99% specificity levels, for the three datasets considered (Truven, UCM and MAYO). For held-back participants in the Truven dataset, ZCoR-IPF sensitivity at 95% specificity was approximately 52% ± 1% for males and 60% ± 2% for females. At 99% specificity, the sensitivity for males fell to 36% ± 2% and to 44% ± 1% for females. In the UCM dataset, we achieved higher performance: 68% ± 14% and 83% ± 13% sensitivities for male and female subsets, respectively, at 95% specificity, and the corresponding values at 99% specificity were 61% ± 16% and 74% ± 13%, respectively. The performance on the MAYO dataset at 95% and 99% specificities was 52% ± 3% and 30% ± 3%, respectively, and is thus closer to the Truven performance. The AUCs for the three datasets were 88% ± 1 − 89% ± 2% for Truven, 88% ± 7 − 94% ± 6% for UCM and 86% ± 3% for MAYO. The precision–recall or the sensitivity versus PPV plots in Fig. 2b assume a uniform prevalence of 5% to compare different subsets that have widely differing prevalences among the US population. Figure 2c shows the plot between LR− and LR+, and Fig. 2d shows the trade-off between NPV and PPV, where again a uniform prevalence of 5% was assumed to compare across the sub-cohorts.
Importantly, the analysis in Fig. 2a,c is independent of the population prevalence of the target disorder and hence offers a more objective assessment of ZCoR-IPF. In particular, we achieved an LR+ > 30 across all cohorts, while keeping the LR− at under or at 0.7. This implies that a participant flagged by ZCoR-IPF has at least 30 times the odds of being diagnosed with IPF 1 year from the screen, compared to participants who were not administered the ZCoR-IPF test. Similarly, the negative likelihood ratio implies that a participant who is not flagged by ZCoR-IPF has at most 70% the odds of being diagnosed with IPF 1 year from the screen, compared to participants who were not administered ZCoR-IPF. Thus, ZCoR-IPF is highly effective in identifying participants at risk of future IPF diagnoses, with lesser effectiveness in ruling out such diagnoses in participants who in reality have little or no risk. By choosing the operating point for ZCoR-IPF (by selecting the decision threshold as discussed earlier), we can decide to operate at a very high LR+ point, or a very low LR− point to achieve different healthcare policy goals.
Results with the broader target definition (the secondary analysis; Extended Data Fig. 3), which demonstrate that while comparison of the ROC curves showed a small degradation in AUC, the analysis also illustrates that we can still maintain LR + > 30 with LR − ≈ 0.7. Thus, on one hand, a broader target definition (targeting ILD) results in somewhat lower but still largely similar predictive performance (Extended Data Fig. 3), and on the other hand, ZCoR-IPF performance on the IPF-Rx and IPF-Ax subsets (Tables 3 and 4 and Extended Data 4 and 5) is not significantly different from that of the full Truven cohort (performance on the IPF-Ax sub-cohort actually being better, although possibly not significantly so), giving us confidence that coding uncertainties do not significantly impact the results (see additional commentary in the Discussion).
In summary, ZCoR-IPF detects about 52–60 of every 100 participants who will be diagnosed with IPF 1 year in future from the point of screening, if we operate at 95% specificity. In cross-validation on the UCM dataset, this number is 68–83 of 100.
Key risk modulators and the comorbidity spectra
Aiming to isolate the key risk modulators, we estimated the relative importance of the features (Fig. 2e) used by ZCoR-IPF, and concluded that past respiratory disorders maximally contribute to the risk, followed by suspected IPF comorbidities (Extended Data 2), metabolic diseases, cardiovascular abnormalities and diseases of the eye. Infections also featured in the top 20 comorbidities. Importantly, despite some differences, the overall pattern of the importance ranking remained substantially invariant across the sexes. We also investigated the statistically significant log-odds ratio of individual ICD codes occurring in the true positive versus the true negative participant sets, which we call the comorbidity spectra (Extended Data Fig. 4), which expectedly showed overrepresentation of circulatory and respiratory disorders. Thus, the patterns we find are in general not surprising; the contribution of this study is to bring them together systematically to realize an accurate risk estimate.
Increase in survival times
Early IPF diagnoses would lead to an immediate increase in post-diagnosis survival times in the population, and we estimated the expected change in survival via a Kaplan–Meier analysis31 (Extended Data Fig. 5). The survival plots (Extended Data Fig. 5a,b) represent lower bounds on the survival function, and upper bounds on the hazard rate, illustrating improved mean survival time from around 100 weeks to 180–200 weeks (shown along with 95% CIs). Importantly, we did not take into account the possibility of prolonging life via clinical interventions after an earlier diagnosis, and thus the actual impact has the potential to be higher. The survival functions are notably similar across the two sexes.
The predictive performance expectedly degraded, as predicted earlier (Extended Data Fig. 5e). Importantly, the degradation was slow enough that we could use ZCoR-IPF with acceptable reliability to predict diagnoses up to 4 years earlier. To illustrate how the ZCoR-IPF risk varies over participant age, we estimated the distribution of the scores over the positive and the control cohorts (Extended Data Fig. 5d). Note that for the participants who eventually get diagnosed, the risk almost linearly increases with age.
Performance in high-risk and low-risk sub-cohorts
We found that our performance (Tables 3 and 4 and Extended Data 4 and 5) was more or less comparable in sub-cohorts defined by a history of diagnoses deemed to be high-risk comorbidities (Extended Data 2). The AUCs achieved for the low-risk cohort were marginally lower (>85% for males and females). Thus, even within the low-risk participants, we still detected 47–50 of every 100 who would be diagnosed in 1 year (Truven), with the corresponding numbers for UCM being 63–74 of 100.
Certainty of idiopathic pulmonary fibrosis diagnosis in participants with target codes (IPF-Rx and IPF-Ax cohorts)
Given that the presence of a target code (Extended Data 1) might sometimes only indicate a suspected diagnosis to be amended later28, and that we did not have pulmonary imaging and other confirmatory test results for our large de-identified databases, we evaluated ZCoR-IPF performance on sub-cohorts in which we can be more confident of the disease state. Two such sub-cohorts are: (1) the ‘IPF-Rx subset’, where the set of positive participants is restricted to those with pirfenidone32 and/or nintedanib33 prescriptions. This additional criterion identifies these participants as having IPF with high certainty (nintedanib is also prescribed in adenocarcinoma of the lungs, but we verified absence of corresponding diagnostic codes to define the IPF-Rx sub-cohort); and (2) ‘the IPF-Ax subset’, where positive participants are obtained by applying the steps enumerated recently in the literature28, namely, (a) age > 50 years, (b) at least two IPF target codes identified at least 1 month apart (codes from the narrow subset in Extended Data 1), (c) a chest CT procedure (ICD-9-CM 87.41 and Current Procedural Terminology, 4th Edition, codes 71250, 71260 and 71270) before the first diagnostic claim for IPF and (d) no claims for alternative ILD codes occurring on or after the first IPF claim (Exclusionary ICD-9 codes for alternative ILD diagnoses: 135, 237.7, 272.7, 277.3, 277.8, 446.21, 446.4, 495, 500 - 505, 506.4, 508.1, 508.8, 516.0, 516.1, 516.32–516.37, 516.2, 516.8, 516.9, 517.0, 517.2, 517.8, 518.3, 555, 710.0, 710.0–710.4, 714.0, 714.81, 720, and 759.5, and their ICD-10 equivalents). In both of these subsets, ZCoR-IPF maintained high performance (Tables 3 and 4 and Extended Data 4 and 5), actually having a better performance on the IPF-Ax subset compared to the full cohorts in the UCM and Truven datasets. Prescriptions of pirfenidone and nintedanib were too few to obtain a meaningfully sized IPF-Rx subset of the UCM dataset.
Finally, we conclude from ablation studies and random shuffles in the participant histories that longitudinal patterns inferred by ZCoR-IPF are demonstrably important for robust performance (Methods).
Discussion
In this study, we develop and cross-validate ZCoR-IPF for IPF screening. Our key finding is that in both men and women aged 45–90 years, ZCoR-IPF accurately identifies IPF cases 1–4 years sooner than occurred in a variety of practice settings during 2003–2021, with clinically relevant predictive performance.
Importantly, ZCoR-IPF achieves such results noninvasively, inexpensively and almost instantaneously, because it relies only on diagnostic data already in the participant’s electronic medical record, and runs on existing information technology infrastructure. The score reflects a sophisticated, highly detailed automated analysis of comorbidities, considering more than 667 features related to the incidence and timing of individual diagnostic codes. ZCoR-IPF thus supplements information currently used to diagnose IPF, namely, respiratory signs and symptoms, pulmonary function and the radiographic and histologic appearance of the lung1,2,34.
Our central claim in this study is the potential utility of ZCoR-IPF as a screening tool in the primary care setting, one that is deployable with little or no additional resources, thus improving delayed and missed diagnosis rates. Importantly, the goal of ZCoR-IPF in this setting is to flag participants for detailed diagnostic evaluation, and not to deliver a final diagnosis by itself.
Thus, ZCoR-IPF can aid primary care physicians to more selectively flag participants for referral for HRCT or to a pulmonologist. Presently, only high-risk participants are flagged, that is, participants with one or more of chronic dyspnea, chronic cough and/or chronic ‘Velcro crackles’ on auscultation, restrictive ventilatory patterns on pulmonary function tests or incidental ILAs or ILD on chest or abdominal CT5,8,12,16,17,18,20,35. As these are relatively large groups, ZCoR-IPF might be applied to help distinguish individuals who require immediate referral versus those who require increased surveillance, versus those who require less frequent follow-up. ZCoR-IPF can be an especially useful tool in participants with ILA, because although these findings might reflect an early stage of the disease, only some 0.5–2% of this group will ever develop IPF8,36.
Additionally, ZCoR-IPF can serve as a diagnostic aid for pulmonologists, radiologists, pathologists or multidisciplinary teams in cases showing abnormalities suggestive of, or associated with IPF, but not UIP on HRCT or histopathology. These cases are relatively frequent: roughly half of participants histopathologically diagnosed with IPF lack classic CT findings associated with the disease8. Hence, ZCoR-IPF might help individuals without UIP on HRCT to avoid more invasive tests, especially surgical lung biopsy2,37, and/or may increase clinicians’ diagnostic confidence; however, such effectiveness needs to be demonstrated prospectively in deployment.
ZCoR-IPF also might speed recruitment and decrease costs of clinical trials of new therapies for IPF and other progressive fibrosing ILD, by allowing more confident inclusion in study samples.
Early screening can also improve efficiency of healthcare resource utilization, via earlier potential access to anti-fibrotic treatment, which might slow the rate of degradation of forced vital capacity in some participants38,39,40,41. Even with traditional therapy, earlier diagnosis triggered by the primary care ZCoR-IPF screen will imply that participants who degrade faster without anti-fibrotic treatment will be identified earlier and hence go on to receive anti-fibrotic medications earlier.
Conversely, earlier diagnosis can avoid unnecessary or harmful treatments, for example, corticosteroids and healthcare visits1,9,10,11,12,13,14,15,42, and might allow more prompt referral for lung transplantation, the only current cure for the disease. Such referral is recommended immediately upon IPF diagnosis, because evaluation for eligibility and waiting times for graft availability may take months or years; starting this process when younger and less ill might allow participants to avoid disqualification for advanced age or frailty1,13. In the meanwhile, quicker IPF diagnosis will accelerate participants’ access to interventions that may improve lung function and quality of life, namely, supplemental oxygen, pulmonary rehabilitation and palliative care13,17, as well as to clinical trials.
Leveraging comorbidities for screening aligns well with the long-standing appreciation of multi-morbidity in IPF24. Indeed, a chart review at a German tertiary referral center43 found 58% of participants with IPF to have 1–3 comorbidities, 30% to have 4–7 comorbidities and only 12% to have no concomitant illness. Understandably some comorbid conditions may be induced by IPF symptoms, for example, depression, and some might represent common misdiagnoses in participants with IPF, for example, COPD, asthma or pneumonia13. Nevertheless, the literature is missing any studies where IPF screening has been attempted from comorbidities alone. The current approaches to risk prediction in IPF are suboptimal44, with predictor models relying on age, sex and pulmonary function, for example, forced vital capacity45, walk tests and lung imaging46, and more recently, specific blood biomarkers47. These models aim to predict the post-diagnosis survival time and yet are moderately accurate at best48,49,50. We lack an approach where future risk is estimated before obvious indications surface—making ZCoR-IPF the potentially first operational tool of its kind.
Universal applicability will require robustness to local variations of participant characteristics. As noted earlier, compared to the Truven dataset, UCM has an overrepresentation of participants under 65 years, and a lower prevalence of high-risk disorders. ZCoR-IPF remains demonstrably unaffected by such local variations, while the performance of the baseline and the NN models on the UCM dataset degrades substantially, making out-of-the-box application of ZCoR-IPF to local populations more reliable, which we found to be lacking for the off-the-shelf tools we investigated.
Despite its promise, less transparent causal links between risk factors and outcome might hinder widespread adoption for ZCoR-IPF. This is a recognized issue with artificial intelligence tools51. However, ZCoR-IPF is not a ‘black box’, with feature importances and the comorbidity spectra delineating diagnoses that maximally modulate future risk. And, since ZCoR-IPF decisions will be followed by confirmatory diagnostic tests, some lack of transparency might be acceptable if meaningful improvements in clinical outcomes are achieved52. Actual deployment of ZCoR-IPF within existing EHR systems is expected to be straightforward, especially after the first EHR-integrated prototype is released as a licensable application in the near future.
A limitation of this study is the use of administrative claims databases without participant-level case validation, implying that some cases of ILD might be prematurely coded as IPF53,54. However, the impact on care might be less concerning, due to (1) ZCoR-IPF being a screening rather than a diagnostic tool, and (2) the emerging concept of ‘progressive chronic fibrosing ILD’ suggesting that subgroups of participants with either IPF or other ILD, for example, idiopathic nonspecific interstitial pneumonia, hypersensitivity pneumonitis, systemic sclerosis-associated ILD or rheumatoid arthritis-associated ILD, may have similar phenotypes and disease behavior, and should perhaps be managed similarly, with early initiation of anti-fibrotic therapy3,55,56. Additionally, comparable performance in our primary and secondary analyses, and in the sub-cohorts designed to more accurately capture the IPF-positive cases (IPF-Rx and IPF-Ax) indicates that coding errors, on average, are not substantially affecting our results.
Another issue with using a commercial claims database for training is potential bias due to older and sicker participants migrating from commercial to government insurance plans57, and potentially limited information on diseases that are insufficiently severe to be considered a discrete diagnosis, for example, dyspnea and cough. Additionally, we did not consider lifestyle variables such as smoking, alcohol consumption, exercise or diet57. Such information might improve our predictions, at the cost of making universal screening at primary care deployments more difficult. This is a design choice differentiating the present study from past attempts at similar artificial intelligence applications58, which typically use age, gender, race, diagnoses, laboratory tests and clinical notes explicitly.
Thus, while claims on accuracy and confidence must be verified prospectively, ZCoR-IPF is a new tool for IPF screening, deployable universally with near-zero drain on healthcare resource, noninvasively, near-instantaneously and repeatedly at the point of care, with the potential for substantial improvement in participant outcomes, participant experience and physician productivity.
Methods
Individual diagnostic histories can have long-term memory59, implying that the order, frequency and comorbid interactions between diseases are important for assessing the future risk of our target phenotype. We analyzed participant-specific diagnostic code sequences by first representing the medical history of each participant as a set of stochastic categorical time series (one each for a specific group of related disorders) followed by the inference of stochastic models for these individual data streams. These inferred generators are from a special class of HMMs, referred to as PFSA26. The inference algorithm we used is distinct from classical HMM learning, and has important advantages related to its ability to infer structure, and its sample complexity. We inferred a separate class of models for the positive and control cohorts, and then the problem reduces to determining the probability that the short diagnostic history from a new participant arises from the positive as opposed to the control category of the inferred models.
Data characteristics
The age-wise breakdown of the cohorts in the Truven and UCM datasets are shown in Supplementary Tables 7 and 8, respectively. The number of diagnostic codes appearing in the Truven dataset is shown in Supplementary Table 9. The Truven dataset represents a national database, and the characteristics of participants in the UCM dataset, comprising participants primarily from Cook County and adjoining areas in the state of Illinois, are somewhat different. In the Truven dataset, we considered approximately 42 million diagnostic codes (with over 46,000 unique codes) for both sexes (Supplementary Table 9) in this analysis, and identified n = 2,053,277 participants, with 53,317 participants in the positive group and 1,999,960 participants in the control group.
Step 1: partitioning the human disease spectrum
We begin by partitioning the human disease spectrum into 51 nonoverlapping broad diagnostic categories. Some of the categories that we define comprise a relatively large number of diagnostic codes aligning roughly with the categories defined within the ICD-9 framework25. The remaining categories represent groups of one or more codes that might have some known or suspected association with pulmonary disorders (Supplementary Table 3 enumerates the categories used in this study). In total, we used 17,008,378 and 25,074,255 diagnostic codes for males and females, respectively (22,685 and 23,722 unique codes), spanning both ICD-9 and ICD-10 protocols (using ICD-10 General Equivalence Mappings60 equivalents where necessary), from a total of 2,053,277 participants. Transforming the diagnostic histories to report only the broad categories reduces the number of distinct codes that the pipeline needs to handle, improving statistical power. Note that defining these categories does not preselect ‘high-risk’ phenotypes; we want our algorithm to seek out the important patterns without any manual curation of the input data.
For each participant, the past medical history is a sequence (t1, x1), ⋯ , (tm, xm), where ti are time stamps and xi are ICD-9 codes diagnosed at time ti. We map individual participant history to a three-alphabet categorical time series zk corresponding to the disease category k, as follows. For each week i, we have the following equation (1):
The time series zk is observed in the inference period. Thus, each participant is represented by 51 trinary series.
Step 2: model inference and computing the sequence likelihood defect Δ
The mapped series are considered to be independent sample paths, and we want to explicitly model these systems as specialized HMMs (PFSAs). The use of PFSA models, along with a measure of divergence between such models known as the SLD, has been demonstrated to achieve high predictive performance, often superseding state-of-the-art frameworks, in multiple applications ranging from general classification problems to incidence forecasts during the coronavirus disease 2019 pandemic to questionnaire-free tools for autism screening27,61,62. We model the positive and the control cohorts and each disease category separately, ending up with 204 HMMs at the population level (51 categories, 2 IPF status categories: positive and control and two sexes). Each of these inferred models is a PFSA, a directed graph with probability-weighted edges, and acts as an optimal generator of the stochastic process driving the sequential appearance of the three letters (as defined by equation (1)) corresponding to disease category, and IPF status type. Structurally, PFSA models are substantially more compact, with the number of model parameters up to several orders of magnitude smaller compared to state-of-the-art NN models (Supplementary Table 5). This, we believe, contributes to ZCoR-IPF’s observed performance and robustness advantages over NN or deep learning models.
To reliably infer the IPF status type of a new participant, that is, the likelihood of a diagnostic sequence being generated by the corresponding IPF status-type model, we use the SLD, which generalizes the notion of Kullback–Leibler divergence63,64 between probability distributions to a divergence \({{{{\mathcal{D}}}}}_{{{{\rm{KL}}}}}(G| | H)\) between ergodic stationary categorical stochastic processes65G,H given by equation (2):
where ∣x∣ is the sequence length, and pG(x), pH(x) are the probabilities of sequence x being generated by the processes G,H, respectively. Defining the log-likelihood of x being generated by a process G according to equation (3):
The cohort type for an observed sequence x (which is actually generated by the hidden process G) can be formally inferred from observations based on the following relationships given by equations (4a) and (4b)27,62:
where \({{{\mathcal{H}}}}(\cdot )\) is the entropy rate of a process63. Equation (4) shows that the computed likelihood has an additional nonnegative contribution from the divergence term when we choose the incorrect generative process. Thus, if a participant is eventually going to be diagnosed with IPF, then we expect that the disease-specific mapped series corresponding to the participant’s diagnostic history be modeled by the PFSA in the positive cohort. Denoting the PFSA corresponding to disease category j for positive and control cohorts as \({G}_{+}^{j},{G}_{0}^{j}\) respectively, we can compute the SLD Δj according to equation (5):
With the inferred PFSA models and the individual diagnostic history, we estimate the SLD on the right-hand side of equation (5). The higher this likelihood defect, the higher the similarity of diagnosis history to that of women with IPF.
The SLD with respect to each broad category is referred to as a PFSA score. In addition to the phenotype-specific PFSA models, we used a range of engineered features that reflect various aspects of the participant-specific diagnostic histories, categorized as the ‘sequence features’, prevalence scores (‘p-scores’) and ‘rare scores’ (see Extended Data 3 for complete list of such features).
Prevalence scores
The p-scores focus on individual diagnostic codes, and we created a dictionary of the ratio of relative prevalence of each code (relative to the set of all codes present) in the positive category (for each sex) to the control category. This is the second hyper-training step. In the later steps of the pipeline, we used dictionary look-ups to map codes to their p-scores, and also their aggregate measures such as mean, median and variance to train a downstream Light Gradient Boosting Machine (LGBM).
Rare scores
These scores consist of a subset of p-scores that correspond to codes with particularly high and low relative prevalences (p-score > 2 or < 0.5). Thus, this feature category depends on the p-score dictionary generated in the second hyper-training step.
Sequence scores
Sequence scores are relatively straightforward statistical measures such as mean, median, variance, time since last occurrence and so on, on the trinary phenotype-specific sex-stratified histories. No hyper-training is required for the generation of the sequence features.
Data splits: training and validation hold-out
To learn the complete set of 667 features, we required three splits of the training dataset. First, all eligible participants were randomly split into the training set (≈75% of data) and the test set (≈25% of data). The training set wss then split into three subsets: (1) the hyper-training set (Supplementary Fig. 2) was used to train PFSA models and the p-score dictionary; (2) the second split (referred to as the pre-aggregation split; Supplementary Fig. 2) was used to train the four feature-category-specific LGBMs; and (3) the final split (referred to as the aggregation split; Supplementary Fig. 2) was used to train the aggregate LGBM, which uses outputs from the trained LGBMs in the previous layer. This trained pipeline was then validated on the held-out validation split (≈25% of data). Figure 2c shows the top 20 features ranked in order of their relative importance (relative loss in performance when dropped out of the analysis).
Step 3: risk estimation pipeline with semi-supervised and supervised learning modules
The SLD along with a range of other engineered features, all functions of data available at the point of care (discussed below) were used to train a network of standard gradient boosting classifiers66 aiming to compute the ZCoR-IPF score.
Statistical analysis
We estimated 95% CIs for all reported metrics. The CIs for the AUC were calculated using the well-known equivalence of the AUC with the Mann–Whitney U statistic67,68,69. We also carried out bootstrapped runs over randomly selected sub-cohorts estimating the distribution of AUC over these runs; the mean AUCs obtained by this approach were within ± 1% of the U-statistic estimate for sufficiently large sub-cohorts. The CIs for specificity and sensitivity were computed via the asymptotic method for single proportions70 (also known as the Wald method). CIs for the remaining metrics were computed from the extremal values of the CI for specificity and sensitivity. We also computed P values for the null hypotheses that ZCoR-IPF performance is different from a corresponding baseline or NN model for the same sex and dataset. The performance difference in the case where the NN model ALEXNET marginally outperformed ZCoR-IPF was not significant; in almost all other cases (only exception being DENSENET for males applied to the Truven dataset), ZCoR-IPF outperformed the corresponding model significantly (Supplementary Table 10). These P values were computed using Dantzig’s upper bound on AUC variance71,72.
Confidence bounds on sensitivity, specificity, positive and negative predictive values and likelihood ratios
The simple asymptotic/Wald method70,73 without continuity correction is sufficient here because the number of samples is large. Thus, for CIs for a ratio of interest p (which in this context can be either specificity or sensitivity), we used equation (6):
where z is the 1 − α/2 point of the standard normal distribution. Note that Newcombe has collated at least seven different approaches to two-sided CIs for quantities that are essentially proportions (such as sensitivity, which is the ratio of true positives to all positives, and specificity, which is the ratio of true negatives to all negatives), including the Wilson score, continuity correction to the asymptotic estimates, methods using exact binomial tail areas and likelihood-based approaches73. The Wald method is the simplest of these, and more sophisticated approaches are called for when the normality assumption cannot be relied upon due to small sample sizes. We verified that these more complex approaches only add minute and insignificant corrections in our case.
Thus, since sensitivity is a ratio involving the number of participants in the positive category, given the empirically estimated sensitivity s, its confidence intervals are computed according to equation (7):
where npositive is the number of participants in the positive category, where as before, z is the 1 − α/2 point of the standard normal distribution.
Similarly, given the empirically estimated specificity c, its CIs are computed according to equation (8):
where ncontrol is the number of participants in the control category. Once the confidence bounds on sensitivity and specificity are determined, the confidence bounds on the remaining quantities are determined using the extremal values of sensitivity and specificity.
Confidence bounds on the area under the curve
The confidence intervals on the estimated AUC values are calculated from the equivalence of the area under the ROC curve with the Mann–Whitney U statistic67,68,69,71,74. Let X and Y be independent random variables, and let x1, x2, ⋯ , xi, ⋯ , xm and y1, y2, ⋯ , yi, ⋯ , ym be samples of X, Y, respectively. The U statistic is defined by equation (9):
It is known67 that the probability measure given by equation (10):
can be estimated by the statistic given by equation (11):
Also it is easily shown that ρ is identical to AUC, and that under realistic assumptions, we have the following, given by equation (12)71:
and that \(\hat{\rho }\) is an unbiased and consistent estimate of ρ. These results allow us to estimate CIs for AUC values at any given significance α, by computing those intervals for \(\frac{U}{mn}\), essentially either by using the variance upper bound given above, or via more sophisticated reasoning where some of the assumptions in equation (12) are not satisfied67,71. In this study, we used the CIs for the U statistic computed by the scipy statistical toolbox for Python3.x.
Approach to comparison with the baseline model
We investigated if ZCoR-IPF outperforms standard or classical approaches, which typically have limited or no pattern discovery. To that effect, we trained and validated a baseline model that considers a broad set of fixed risk factors as binary features (recording presence or absence in participant history within past 2 years) and finally uses standard logistic regression to train a classifier predicting an IPF diagnosis (appearance of one of the target codes) 1 year in future. The 87 diagnostic codes defining the binary features used in this baseline model are enumerated in Supplementary Table 4, which relate to asthma and COPD-associated major codes, cough, dyspnea and other major pulmonary and cardiopulmonary complications. The baseline model also uses age as a feature (recording if participants are 65 years or older).
Approach to comparing with neural network architectures
To adequately compare the performance of NNs to our model, we used the same input data for each participant as we described in ‘ZCoR-IPF modeling, training and prediction’. To infer the risk score, we looked at 2 years of diagnostic records preceding the date of screening. As a result, each participant is represented by 51 104-week-long sparse stochastic time series of events corresponding to 51 different disease groups, each having 104 ternary digits for each week: ‘1’ if a disease from a given disease group was diagnosed that week, ‘2’ if any other disease was diagnosed that week and ‘0’ if no diseases were diagnosed that week. If no codes were recoded for a certain time series for the whole window of observation, all of its digits were marked as ‘−1’. For each participant, a list of 51 104-week-long time series is transformed into the 51 × 104 matrix, where time-series rows appear in the same order as they appear in inputs for training ZCoR-IPF (Supplementary Fig. 3).
We investigated the predictive performance achieved by diverse NN and standard deep learning architectures, ranging from simple feed-forward networks75 to long short-term memory (LSTM) models76, convolutional neural networks (CNNs77) to more advanced ALEXNET78,79, DENSENET80 and RESNET81, RETAIN82 and multichannel convolutional architectures (Supplementary Note). LSTM networks were chosen to investigate the impact of the ability of these architectures to model long-range longitudinal dependencies. CNNs were chosen to test their ability to leverage patterns and interdependencies across concurrent time series of different disease groups. The more standard architectures were investigated to evaluate the ability of past successful NN models in this context. The RETAIN architecture was investigated for its claimed optimized yet interpretable structure specifically targeting the health domain. Similar to the ZCoR-IPF approach, we trained and tested separate models for male and female cohorts.
We trained these networks to deliver the maximum out-of-sample performance on the Truven dataset (with similar test/train data splits as used for ZCoR-IPF) and recorded their performance on the UCM validation dataset. Similarly to ZCoR-IPF, we trained and tested separate models for male and female cohorts.
The performance of simpler architectures did not produce good results, irrespective of whether we used LSTMs or CNNs or modifications thereof. Multichannel architectures, for example, CNN_MULTICHANNEL, were also investigated to explore if performance enhancement is obtained from using 51 separate 104-digit-long one-dimensional arrays for 51 parallel sets of convolutional layers (instead of a 51 × 104 two-dimensional matrix as input) that are concatenated for the final set of dense layers that provide the final prediction. A summary of the different NN models we investigated is shown in Supplementary Table 5.
Our results suggest that simple NN architectures are substantially worse than ZCoR-IPF, but are generally better than the baseline model (Extended Data Fig. 2), while typically having larger model complexity. Exploring state-of-the-art deep NN models (Supplementary Table 5), we were able to train and validate the ALEXNET architecture to outperform ZCoR-IPF on out-of-sample data in the Truven dataset (ALEXNET, 94% vs ZCoR-IPF, 89% AUC). However, the subsequent performance of the trained ALEXNET on the UCM dataset was substantially worse (ALEXNET, 69% vs ZCoR-IPF, 93% AUC). At the same time, the ALEXNET model with 22,616,129 parameters was three orders of magnitude more complex compared to ZCoR-IPF with 64,091 parameters. We calculated P values for the significance of ZCoR-IPF outperforming (Supplementary Table 10) and show that (1) we significantly outperform competing models, and (2) the case where ALEXNET marginally beats ZCoR-IPF is not statistically significant.
We concluded that some NNs (ALEXNET) can achieve high out-of-sample performance in the Truven database, and at least one architecture (ALEXNET) successfully outperformed the ZCoR-IPF; however, performance of the trained NN pipeline can be significantly worse in an independent database, while the ZCoR-IPF maintains high performance across databases. This pattern seems to hold true for all NN models we experimented with. Note that this is not a simple case of overtraining, which was diligently accounted for in the training phase. Thus, it appears that the differences in participant characteristics between Truven and UCM (Table 2) have a dramatic impact on NN performance, while not affecting the ZCoR-IPF significantly. It is possible some specific encoding of the data can remedy this issue for NN and deep learning models.
Ablation studies and approach to establish the necessity of longitudinal patterns
Among the multiple feature categories, p-scores are designed to capture the impact of individual codes via computing the risk ratio of specific ICD codes in the positive cohorts compared with the control cohorts. The PFSA scores, on the other hand, are designed to capture longitudinal patterns in and across broad phenotypic categories described before. It is important to inquire if the added complexity of recognizing longitudinal patterns is at all necessary, that is, do the patterns that modulate risk have any longitudinal dependencies. We investigate this question in two ways. First, we consider ablation studies, where we, on one hand, train pipelines using only PFSA-based features and no p-scores and, on the other hand, train using only p-score-based features and no PFSA-based ones, thus eliminating longitudinal patterns. Second, we randomly permute the encounter time stamps for individual participants in the testing and validation stages and evaluate if such shuffled histories impact prediction decisions made by the ZCoR-IPF pipeline.
These interrogations show that longitudinal patterns inferred by ZCoR-IPF are key to maintaining high performance under noise and uncertainties (Supplementary Fig. 1). Our ablation studies on the different feature categories used by ZCoR-IPF show that the p-score by itself (without the PFSA-based features, and therefore having no longitudinal pattern discovery capability) does better in some scenarios than when using PFSA-based features alone. Nevertheless, the combined models perform better than either, suggesting that longitudinal patterns have quantifiable predictive value. Additionally, when we perturb participant histories by randomly swapping diagnostic codes with one representing similar or confounding disorders (Supplementary Table 11), p-scores alone have quite dismal performance, and the performance is recovered when we use PFSA-based features in combination with p-scores (Supplementary Fig. 1). This trend is replicated with the full cohort in the Truven dataset, as well as the high-risk and the low-risk sub-cohorts. We also evaluated the scenario where longitudinal patterns existing in the data are erased by randomly permuting the recorded time stamps in individual participant histories. When the out-of-sample validation cohort in the Truven dataset was permuted in this manner, we observed (Supplementary Fig. 1) that at 95% specificity, ZCoR-IPF decisions switched in 10% of the participants who were predicted to be in the positive cohort with original unperturbed histories. At 90% specificity, this fraction increases to 15%. Similarly, predictions switched from the control to the positive category in 0.5% of the cases at 95% specificity, and in 0.75% of the cases at 90% specificity. These investigations demonstrate that longitudinal patterns are indeed predictive of future IPF risk.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The Truven, UCM and MAYO datasets cannot be made available due to their commercial nature. D.O. and I.C. had access to the Truven and UCM databases, and I.C. was responsible for maintaining the integrity of these datasets. C.G.N., L.J.F. and A.H.L. had access to the MAYO dataset, and A.H.L. was responsible for maintaining the integrity of that dataset.
Code availability
Methodological details needed to evaluate our conclusions are included in the Methods and Supplementary Information. A working software implementation of the pipeline (free for noncommercial evaluations) is available at https://doi.org/10.5281/zenodo.6040418, which includes installation instructions in standard Python environments. To enable fast execution, some more compute-intensive features are disabled in this version. Results from this software are for demonstration purposes only, and must not be interpreted as medical advice, or serve as replacement for such.
References
Lederer, D. & Martinez, F. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 378, 1811–1823 (2018).
Raghu, G., Remy-Jardin, M. & Myers, J. Diagnosis of idiopathic pulmonary fibrosis. an official ats/ers/jrs/alat clinical practice guideline. Am. J. Respir. Crit. Care Med. 198, 44–68 (2018).
Raghu, G. Idiopathic pulmonary fibrosis: shifting the concept to irreversible pulmonary fibrosis of many entities. Lancet Respir. Med. 7, 926–929 (2019).
Ley, B., Collard, H. & King, T., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 183, 431–440 (2011).
Antoniou, K., Symvoulakis, E., Margaritopoulos, G., Lionis, C. & Wells, A. Early diagnosis of IPF: time for a primary-care case-finding initiative? Lancet Respir. Med. 2, 1 (2014).
Adegunsoye, A. Diagnostic delay in idiopathic pulmonary fibrosis: where the rubber meets the road. Ann. Am. Thorac. Soc. 16, 310–312 (2019).
Cottin, V. & Richeldi, L. Neglected evidence in idiopathic pulmonary fibrosis and the importance of early diagnosis and treatment. Eur. Respir. Rev. 23, 106–110 (2014).
Putman, R., Rosas, I. & Hunninghake, G. Genetics and early detection in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 189, 770–778 (2014).
Lamas, D. et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am. J. Respir. Crit. Care Med. 184, 842–847 (2011).
Hoyer, N., Prior, T., Bendstrup, E., Wilcke, T. & Shaker, S. Risk factors for diagnostic delay in idiopathic pulmonary fibrosis. Respir. Res. 20, 103 (2019).
Mooney, J., Chang, E. & Lalla, D. Potential delays in diagnosis of idiopathic pulmonary fibrosis in medicare beneficiaries. Ann. Am. Thorac. Soc. 16, 393–396 (2019).
Pritchard, D., Adegunsoye, A. & Lafond, E. Diagnostic test interpretation and referral delay in patients with interstitial lung disease. Respir. Res. 20, 253 (2019).
Cosgrove, G. P., Bianchi, P., Danese, S. & Lederer, D. J. Barriers to timely diagnosis of interstitial lung disease in the real world: the INTENSITY survey. BMC Pulm. Med. 18, 9 (2018).
Schoenheit, G., Becattelli, I. & Cohen, A. Living with idiopathic pulmonary fibrosis: an in-depth qualitative survey of European patients. Chron. Respir. Dis. 8, 225–231 (2011).
Collard, H., Tino, G. & Noble, P. Patient experiences with pulmonary fibrosis. Respir. Med. 101, 1350–1354 (2007).
Thickett, D., Voorham, J. & Ryan, R. Historical database cohort study addressing the clinical patterns prior to idiopathic pulmonary fibrosis (IPF) diagnosis in UK primary care. BMJ Open 10, 034428 (2020).
Hewson, T. et al. Timing of onset of symptoms in people with idiopathic pulmonary fibrosis. Thorax https://doi.org/10.1136/thoraxjnl-2017-210177 (2017).
Cottin, V. & Cordier, J. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur. Respir. J. 40, 519–521 (2012).
Hart, S. Machine learning molecular classification in IPF: UIP or not UIP, that is the question. Lancet Respir. Med. 7, 466–467 (2019).
Oldham, J. & Noth, I. Idiopathic pulmonary fibrosis: early detection and referral. Respir. Med. 108, 819–829 (2014).
Hansen, L. The Truven Health MarketScan Databases for Life Sciences Researchers (Truven Health Ananlytics IBM Watson Health, 2017).
Andrade, C. Examination of participant flow in the CONSORT diagram can improve the understanding of the generalizability of study results. J. Clin. Psychiatry 76, e1469–e1471 (2015).
Wallace, P. J., Shah, N. D., Dennen, T., Bleicher, P. A. & Crown, W. H. Optum Labs: building a novel node in the learning healthcare system. Health Aff. 33, 1187–1194 (2014).
Raghu, G., Amatto, V., Behr, J. & Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur. Respir. J. 46, 1113–1130 (2015).
World Health Organization. International Classification of Diseases—Ninth Revision (ICD-9). Wkly Epidemiol. Rec. 63, 343–344 (1988).
Chattopadhyay, I. & Lipson, H. Abductive learning of quantized stochastic processes with probabilistic finite automata. Philos. Trans. A Math. Phys. Eng. Sci. 371, 20110543 (2013).
Huang, Y. & Chattopadhyay, I. Universal risk phenotype of us counties for flu-like transmission to improve county-specific covid-19 incidence forecasts. PLoS Comput. Biol. 17, e1009363 (2021).
Ley, B. et al. Code-based diagnostic algorithms for idiopathic pulmonary fibrosis. Case validation and improvement. Ann. Am. Thorac. Soc. 14, 880–887 (2017).
Alqarni, A. M., Schneiders, A. G. & Hendrick, P. A. Clinical tests to diagnose lumbar segmental instability: a systematic review. J. Orthop. Sports Phys. Ther. 41, 130–140 (2011).
Vining, R., Potocki, E., Seidman, M. & Morgenthal, A. P. An evidence-based diagnostic classification system for low back pain. J. Can. Chiropr. Assoc. 57, 189–204 (2013).
Kaplan, E. L. & Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 53, 457–481 (1958).
Noble, P. W. et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (capacity): two randomised trials. Lancet 377, 1760–1769 (2011).
Richeldi, L. et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2071–2082 (2014).
Hyldgaard, C., Hilberg, O. & Bendstrup, E. How does comorbidity influence survival in idiopathic pulmonary fibrosis? Respir. Med. 108, 647–653 (2014).
Oldham, J., Adegunsoye, A. & Khera, S. Underreporting of interstitial lung abnormalities on lung cancer screening computed tomography. Ann. Am. Thorac. Soc. 15, 764–766 (2018).
Walsh, S., Humphries, S., Wells, A. & Brown, K. Imaging research in fibrotic lung disease; applying deep learning to unsolved problems. Lancet Respir. Med. 8, 1144–1153 (2020).
Raghu, G., Flaherty, K. & Lederer, D. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir. Med. 7, 487–496 (2019).
Torrisi, S. E., Pavone, M., Vancheri, A. & Vancheri, C. When to start and when to stop antifibrotic therapies. Eur. Respir. Rev. 26, 170053 (2017).
Sugino, K. et al. Efficacy of early antifibrotic treatment for idiopathic pulmonary fibrosis. BMC Pulm. Med. 21, 218 (2021).
Ryerson, C. J. et al. Effects of nintedanib in patients with idiopathic pulmonary fibrosis by gap stage. ERJ Open Res. 5, 00127–2018 (2019).
Kropski, J. Biomarkers and early treatment of idiopathic pulmonary fibrosis. Lancet Respir. Med. 7, 725–727 (2019).
Farrand, E., Iribarren, C. & Vittinghoff, E. Impact of idiopathic pulmonary fibrosis on longitudinal health-care utilization in a community-based cohort of patients. Chest 159, 219–227 (2020).
Kreuter, M., Ehlers-Tenenbaum, S. & Palmowski, K. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS ONE 11, 0151425 (2016).
Ley, B. & Collard, H. R. Risk prediction in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 185, 6–7 (2012).
Ryerson, C. J. et al. Predicting mortality in systemic sclerosis-associated interstitial lung disease using risk prediction models derived from idiopathic pulmonary fibrosis. Chest 148, 1268–1275 (2015).
Kim, G. H. J. et al. Prediction of idiopathic pulmonary fibrosis progression using early quantitative changes on ct imaging for a short term of clinical 18- to 24-month follow-ups. Eur. Radiol. 30, 726–734 (2020).
Richards, T. J. et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 185, 67–76 (2012).
King Jr, T. E., Tooze, J. A., Schwarz, M. I., Brown, K. R. & Cherniack, R. M. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am. J. Respir. Crit. Care Med. 164, 1171–1181 (2001).
Wells, A. U. et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am. J. Respir. Crit. Care Med. 167, 962–969 (2003).
du Bois, R. M. et al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 184, 459–466 (2011).
Singh, R. P., Hom, G. L., Abramoff, M. D., Campbell, J. P. & Chiang, M. F. Current challenges and barriers to real-world artificial intelligence adoption for the healthcare system, provider, and the patient. Transl. Vis. Sci. Technol. 9, 45 (2020).
Holm, E. A. In defense of the black box. Science 364, 26–27 (2019).
Esposito, D., Lanes, S. & Donneyong, M. Idiopathic pulmonary fibrosis in united states automated claims. incidence, prevalence, and algorithm validation. Am. J. Respir. Crit. Care Med. 192, 1200–7 (2015).
Ley, B., Urbania, T. & Husson, G. Code-based diagnostic algorithms for idiopathic pulmonary fibrosis. Case validation and improvement. Ann. Am. Thorac. Soc. 14, 880–887 (2017).
Inoue, Y., Kaner, R. & Guiot, J. Diagnostic and prognostic biomarkers for chronic fibrosing interstitial lung diseases with a progressive phenotype. Chest 158, 646–659 (2020).
George, P., Spagnolo, P. & Kreuter, M. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir. Med. 8, 925–934 (2020).
Mortimer, K., Bartels, D. & Hartmann, N. Characterizing health outcomes in idiopathic pulmonary fibrosis using US health claims data. Respiration 99, 108–118 (2020).
Miotto, R., Li, L., Kidd, B. A. & Dudley, J. T. Deep patient: an unsupervised representation to predict the future of patients from the electronic health records. Sci. Rep. 6, 26094 (2016).
Granger, C. W. J. & Joyeux, R. An introduction to long-memory time series models and fractional differencing. J. Time Ser. Anal. 1, 15–29 (1980).
American Academy of Pediatrics. Transitioning to 10: 2014 general equivalence mappings (online exclusive). AAP Pediatric Coding Newsletter https://doi.org/10.1542/pcco_book116_document005 (2013).
Chattopadhyay, I. & Lipson, H. Data smashing: uncovering lurking order in data. J. R. Soc. Interface 11, 20140826 (2014).
Onishchenko, D. et al. Reduced false positives in autism screening via digital biomarkers inferred from deep comorbidity patterns. Sci. Adv. 7, eabf0354 (2021).
Cover, T. M. & Thomas, J. A. Elements of Information Theory (Wiley-Interscience, 1991).
Kullback, S. & Leibler, R. A. On information and sufficiency. Ann. Math. Statist. 22, 79–86 (1951).
Doob, J. Stochastic Processes (Wiley, 1953). https://books.google.com/books?id=KvJQAAAAMAAJ
Ke, G. et al. LightGBM: a highly efficient gradient boosting decision tree. In Advances in Neural Information Processing Systems 3146–3154 (2017).
Birnbaum, Z. W. & Klose, O. M. Bounds for the variance of the Mann–Whitney statistic. Ann. Math. Stat. 4, 933–945 (1957).
Mann, H. B. & Whitney, D. R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 18, 50–60 (1947).
Wilcoxon, F. Individual comparisons by ranking methods. in Breakthroughs in Statistics Vol. 2 196–202 (Springer, 1992).
Newcombe, R. G. & Vollset, S. E. Confidence intervals for a binomial proportion. Stat. Med. 13, 1283–1285 (1994).
Birnbaum, Z. On a use of the Mann–Whitney statistic. in Contribution to the Theory of Statistics Vol. 1, 13–18 (University of California Press, 2020).
van Dantzig, D. On the consistency and the power of wilcoxon’s two-sample test (Proceedings KNAW series A, 54, nr 1, Indagationes Mathematicae, 13, 1–8). Stichting Mathematisch Centrum. Statistische Afdeling (1951).
Newcombe, R. G. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat. Med. 17, 857–872 (1998).
Haldane, J. B. & Smith, C. A. A simple exact test for birth-order effect. Ann. Eugen. 14, 117–124 (1947).
Schmidhuber, J. Deep learning in neural networks: an overview. Neural Netw. 61, 85–117 (2015).
Van Houdt, G., Mosquera, C. & Nápoles, G. A review on the long short-term memory model. Artif. Intell. Rev. 53, 5929–5955 (2020).
Albawi, S., Mohammed, T. A. & Al-Zawi, S. Understanding of a convolutional neural network. In 2017 International Conference on Engineering and Technology (ICET), 1–6 (IEEE, 2017).
Krizhevsky, A., Sutskever, I. & Hinton, G. E. ImageNet classification with deep convolutional neural networks. Commun. ACM 60, 84–90 (2017).
Alom, M. Z. et al. The history began from AlexNet: a comprehensive survey on deep learning approaches. Preprint at https://arxiv.org/abs/1803.01164 (2018).
Zhang, K., Guo, Y., Wang, X., Yuan, J. & Ding, Q. Multiple feature reweight densenet for image classification. IEEE Access 7, 9872–9880 (2019).
Lu, Z., Jiang, X. & Kot, A. Deep coupled resnet for low-resolution face recognition. IEEE Signal Processing Lett. 25, 526–530 (2018).
Guo, W., Ge, W., Cui, L., Li, H. & Kong, L. An interpretable disease onset predictive model using crossover attention mechanism from electronic health records. IEEE Access 7, 134236–134244 (2019).
Acknowledgements
This work is funded in part by the Defense Advanced Research Projects Agency under project no.HR00111890043. The claims made in this study do not reflect the position or the policy of the US Government. The UCM dataset is provided by the Clinical Research Data Warehouse (CRDW) maintained by the Center for Research Informatics at the University of Chicago. The Center for Research Informatics is funded by the Biological Sciences Division, the Institute for Translational Medicine/CTSA (National Institutes of Health award no. UL1 TR000430) at the University of Chicago.
Author information
Authors and Affiliations
Contributions
D.O. implemented the algorithm and ran validation tests. D.O. and I.C. carried out mathematical modeling and algorithm design. R.J.M., F.J.M. and I.C. wrote the paper. F.J.M., G.M.H. and I.C. interpreted results and guided research. C.G.N., L.J.F. and A.H.L. evaluated the tool on the dataset available at the Mayo Clinic. I.C. procured funding for the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Athol Wells, Harold Collard and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Michael Basson, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Performance under delayed updates to participant records.
a, b, Out-of-sample ROC curves when the patient data is delayed by 4w vs the no-delay condition, for the UCM and the Truven datasets, respectively. 95% confidence bounds about the mean is shown, computed with n=2,053,277 for Truven and n=68,658 for UCM. Note that there is no significant loss of performance with such delayed data. c, d, ZCoR-IPF performance vs a 87-feature baseline model optimized via logistic regression, where these features denote presence/absence of manually-curated risk factors (Supplemental Table 4) and age (over/under 65 years), for the Truven and the UCM datasets, respectively.
Extended Data Fig. 2 Comparison with neural network architectures.
a,b, Out-of-sample AUC achieved in Truven and UCM datasets, respectively, by a range of neural network architectures ranging from simple feed-forward networks, LSTMs and CNNs, to large state of the art models such as the ALEXNET, DENSENET and RESNET, along with 95% confidence intervals about the mean (n=2,053,277 for Truven and n=68,658 for UCM).
Extended Data Fig. 3 Performance with broader target definition.
a,b, Out-of-sample ROC curves for the Truven and the UCM dataset, respectively, comparing the results from the primary analysis with that in the secondary analysis (analysis with broader target definition as specified in Extended Data Table 1). 95% confidence bounds about the mean is shown, computed with n=2,053,277 for Truven and n=68,658 for UCM. c, Negative vs positive likelihood ratios (LR- vs LR+). d, Positive vs negative predictive values. Note that with the broad target definition we can select to operate with LR+ > 30 as well, similar to the target in the primary analysis.
Extended Data Fig. 4 Co-morbidity Spectra.
a,b, Diseases (recorded ICD codes) that increase the odds of the patient being a ‘true positive’ vs a ‘true negative’ for males and females respectively. These odds are broadly similar across the sexes, with over-representation of respiratory disorders.
Extended Data Fig. 5 Expected increase in survival times.
a, Survival function lower bounds at two specificity levels (90 and 95%). b, Cumulative hazard function upper bounds. 95% confidence bounds around the mean shown for both, generated using the Truven dataset (n=2,053,277). c, Variation of the mean survival time as a function of the specificity at which ZCoR-IPF is operated. d, Variation of estimated raw risk as a function of age for screening four years from actual recorded diagnosis of IPF, showing that risk increases almost linearly with age for the patients eventually diagnosed with IPF. e, Degradation of out-of-sample AUC as we attempt to screen earlier, stepping back from the time of current diagnosis (in absence of ZCoR-IPF screening).
Supplementary information
Supplementary Information
Supplementary Note, Tables 1–11 and Figs. 1–3
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Onishchenko, D., Marlowe, R.J., Ngufor, C.G. et al. Screening for idiopathic pulmonary fibrosis using comorbidity signatures in electronic health records. Nat Med 28, 2107–2116 (2022). https://doi.org/10.1038/s41591-022-02010-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-022-02010-y
This article is cited by
-
Epidemiology and comorbidities in idiopathic pulmonary fibrosis: a nationwide cohort study
BMC Pulmonary Medicine (2023)
