
Abstract
Anti-BRAF/EGFR therapy was recently approved for the treatment of metastatic BRAFV600E colorectal cancer (mCRCBRAF-V600E). However, a large fraction of patients do not respond, underscoring the need to identify molecular determinants of treatment response. Using whole-exome sequencing in a discovery cohort of patients with mCRCBRAF-V600E treated with anti-BRAF/EGFR therapy, we found that inactivating mutations in RNF43, a negative regulator of WNT, predict improved response rates and survival outcomes in patients with microsatellite-stable (MSS) tumors. Analysis of an independent validation cohort confirmed the relevance of RNF43 mutations to predicting clinical benefit (72.7% versus 30.8%; P = 0.03), as well as longer progression-free survival (hazard ratio (HR), 0.30; 95% confidence interval (CI), 0.12–0.75; P = 0.01) and overall survival (HR, 0.26; 95% CI, 0.10–0.71; P = 0.008), in patients with MSS-RNF43mutated versus MSS-RNF43wild-type tumors. Microsatellite-instable tumors invariably carried a wild-type-like RNF43 genotype encoding p.G659fs and presented an intermediate response profile. We found no association of RNF43 mutations with patient outcomes in a control cohort of patients with MSS-mCRCBRAF-V600E tumors not exposed to anti-BRAF targeted therapies. Overall, our findings suggest a cross-talk between the MAPK and WNT pathways that may modulate the antitumor activity of anti-BRAF/EGFR therapy and uncover predictive biomarkers to optimize the clinical management of these patients.
Similar content being viewed by others


Main
The criteria to match patients with cancer with the most effective therapies relies on the identification of molecular tumor dependencies that can be targeted with available treatments1. The BRAFV600E mutation is found in approximately 10% of patients with metastatic colorectal cancer (mCRC), and its clinical presentation is often associated with a predominance of right-sided proximal tumors, high prevalence of microsatellite instability (MSI; near 30%), refractoriness to standard-of-care therapies and an unfavorable prognosis (Extended Data Fig. 1a)2,3. Compared to mCRCBRAF-wild-type, BRAFV600E tumors (hereafter referred to as mCRCBRAF-V600E) also associate with specific molecular features, including a low frequency of APC mutations and a high rate of mutations in the tumor suppressor gene RNF43 (refs. 4,5), a RING E3 ubiquitin ligase involved in suppression of the WNT–β-catenin pathway through promoting the degradation of FZD/WNT receptors6,7.
BRAFV600E ATP-competitive kinase inhibitors were designed and clinically tested for the treatment of BRAFV600E-driven tumors, thus achieving variable outcomes depending on tumor type8. In particular, patients with melanoma harboring BRAFV600E mutations have been demonstrated to derive marked benefit from BRAF inhibitor monotherapy (up to 70% objective response rate (ORR)9,10), while, in stark contrast, patients with BRAFV600E-mutant CRC receiving the same treatment experienced little clinical benefit8,11,12. Preclinical studies uncovered an intricate molecular circuitry in CRCBRAF-V600E leading to rapid compensatory activation of the epidermal growth factor receptor (EGFR) that likely hampered the clinical outcomes of these patients13,14. These key findings set the rationale for the design of clinical trials targeting both BRAF and EGFR, with or without additional targeted therapies (that is, MEK, ERK or PIK3CA inhibitors) that generally achieved improved clinical outcomes as compared to previous standard-of-care treatments12,15,16,17.
The clinical outcomes of patients with mCRCBRAF-V600E treated with the triplet regimen of encorafenib, cetuximab and binimetinib were assessed in the phase 3 BEACON CRC trial (NCT02928224). The study showed that combinatorial blockade of BRAF and EGFR, with or without concomitant MEK inhibition, achieved improved ORR (26% with the triplet and 20% with the doublet therapy versus 2% in the control arm) and extended overall survival (OS) and progression-free survival (PFS)18,19. These results warranted approval of the doublet therapy as a new standard therapy for CRCBRAF-V600E by the US Food and Drug Administration and European Medicines Agency20. While the responses documented are unprecedented for patients with mCRCBRAF-V600E, these still compare unfavorably with the higher response rates observed in BRAF-mutant metastatic melanomas treated with anti-BRAF therapy9,10 and display a high degree of heterogeneity, which underscores the need for a deeper understanding of factors modulating treatment response that can optimize the clinical management of patients1,21.
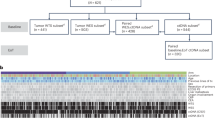
Herein we sought to explore genetic biomarkers with a predictive value that can contribute to refining the stratification of patients with mCRCBRAF-V600E treated with anti-BRAF/EGFR combinatorial therapy. We applied whole-exome sequencing (WES) and/or targeted gene sequencing on baseline tumor and/or plasma cell-free DNA (cfDNA) samples from a large cohort of patients with mCRCBRAF-V600E treated with anti-BRAF/EGFR therapy, as well as from a control cohort of patients with mCRCBRAF-V600E receiving standard chemotherapies and antiangiogenic agents (and not exposed to anti-BRAF), and integrated these data with clinical correlates of response and survival. Our findings identified molecular subtypes based on microsatellite-stable (MSS)/MSI status and RNF43 alterations and uncovered the predictive value of RNF43 mutations as a biomarker of clinical outcome, including increased ORR, PFS and OS, to anti-BRAF/EGFR ± combinatorial therapies. Specifically, our data show that patients with MSS-mCRCBRAF-V600E tumors harboring loss-of-function mutations in RNF43 respond favorably to anti-BRAF/EGFR combinatorial therapy, whereas those with functional RNF43 derived limited benefit from this regimen (Fig. 1).

A total of 166 patients with mCRCBRAF-V600E were included in the study from discovery (n = 46), validation (n = 52) and control (n = 68) cohorts. WES of germline DNA, baseline tumor DNA and/or baseline plasma cfDNA from 28 patients was performed. Targeted NGS was used to assess RNF43 tumor mutation status for the 18 remaining patients from the discovery cohort and all tumors from the validation and control cohorts. Genomic profiles and MSS/MSI-RNF43 molecular subtypes were compared with clinical response data (ORR, mPFS and mOS) using dNdScv maximum-likelihood unbiased mutation enrichment analysis25. In vitro assays were used to assess the functional impact of RNF43 mutations detected in patient samples (see more in Fig. 6).
Results
Description of patient cohorts treated with anti-BRAF/EGFR
A total of 98 patients with mCRCBRAF-V600E treated with anti-BRAF/EGFR ± combinatorial therapies in clinical trials or who received such therapies through a compassionate use program were included in the current study (Fig. 1 and Extended Data Fig. 1b). The discovery cohort was composed of 46 patients from the Vall d’Hebron University Hospital (Barcelona, Spain) prospectively included from 2013 to 2021, and the validation cohort comprised 52 patients from three academic hospitals from Italy (Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; University Hospital of Pisa, Pisa, Italy; Istituto Oncologico Veneto IOV-IRCCS, Padova, Italy).
In the discovery cohort, 28 patients (61%) were female, with a median age at diagnosis of 61 years (range, 33–82 years), 31 patients (67%) had right-sided tumors, 34 patients (74%) had more than one metastatic site, and 37 patients (80%) had MSS and 9 patients (20%) had MSI tumors. In total, 35 patients (76%) received anti-BRAF/EGFR-based combinations as second- or third-line therapy, 6 patients (13%) received this as first-line therapy and 5 patients (11%) received this beyond third-line therapy. The number of patients who received the doublet combination was 29 (63%), and 17 patients (37%) received the triplet combination. The ORR by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1; ref. 22) was 44% (20/45 patients, one patient was not evaluable for response). The number of patients with MSI tumors who received immunotherapy after targeted therapy was 3 (7%). The RNF43 mutation frequency was 43% (20/46) in the overall discovery cohort, 100% (9/9, all G659fs) in MSI tumors and 30% (11/37) in MSS tumors (Supplementary Table 1 and Fig. 2a).

a,b, Waterfall plots representing best observed response in the mCRCBRAF-V600E discovery cohort (n = 44) (a) and validation cohort (n = 42) (b), measured as percentage best change from baseline according to RECIST 1.1 criteria22 (Methods). Colors refer to molecular subtypes: MSS-RNF43wild-type tumors (yellow), MSS-RNF43mutated tumors (blue) and MSI-RNF43mutated tumors (gray).
In the validation cohort, 31 patients (60%) were female, with a median age at diagnosis of 62 years (range, 38–80 years), 40 patients (77%) had right-sided tumors, 33 patients (63%) had more than one metastatic site, and 39 patients (75%) had MSS and 13 patients (25%) had MSI tumors. Overall, 33 (63%) patients received anti-BRAF/EGFR-based combinations as second-line therapy, and 19 patients (37%) received this as third-line therapy. The number of patients who received the doublet combination was 29 (56%), and 23 patients (44%) received the triplet combination. The ORR by RECIST 1.1 (ref. 22) was 27% (14/51 patients, one patient was not evaluable for response). Six patients (12%) with MSI tumors received immunotherapy after targeted therapy. The RNF43 mutation frequency was 44% (23/52) in the overall validation cohort, 92% (12/13, G659fs) in MSI tumors and 28% (11/39) in MSS tumors (Supplementary Table 1 and Fig. 2b).
Biomarkers of response to anti-BRAF/EGFR in MSS-mCRCBRAF-V600E
WES analysis detected mutations in several cancer-related genes previously reported to be implicated in mCRCBRAF-V600E tumors, such as APC, TP53, ARID1A, PIK3CA, FBXW7 and RNF43, among others (Supplementary Table 2). In general, the mutation frequencies of the discovery cohort were comparable to those observed in The Cancer Genome Atlas (TCGA) CRC tumors carrying the BRAFV600E mutation from the PanCancer Atlas23,24 (n = 46; Supplementary Table 2). Unbiased maximum-likelihood analysis of WES mutational data from responders (partial response (PR) and complete response (CR)) versus nonresponders (stable disease (SD) and progressive disease (PD)) using dNdScv25 identified the RNF43 gene as a top candidate gene associated with ORR (P values and q values <0.001; Extended Data Fig. 2a,b). Other candidate genes were identified, but we focused on RNF43 because of its high mutation frequency and implications in CRC biology. RNF43 mutations were detected in both tumor-derived and plasma cfDNA-derived DNA samples (Extended Data Fig. 3a–c).
The mutation rate of RNF43 in the discovery cohort (20/46, 43%) was consistent with the rate reported in the TCGA PanCancer Atlas for CRCBRAF-V600E (refs. 23,24; 21/46, 46%). To test the potential association between RNF43 mutations and ORR, we analyzed data from the whole discovery series including WES and next-generation sequencing (NGS) panel data from 45 evaluable patients with mCRCBRAF-V600E. ORR in the whole RNF43mutated subgroup, including MSS and MSI tumors, was 63% (12/19), which was significantly superior to the ORR of 31% achieved in patients with RNF43wild-type status (8/26; P = 0.03; Extended Data Fig. 4a). The accuracy of RNF43 mutations alone for predicting response to treatment was 67% (sensitivity, 60%; specificity, 72%; positive predictive value (PPV), 63%; negative predictive value (NPV), 69%; Extended Data Fig. 4g). In line with findings in the BEACON trial, patients with MSS and MSI CRCBRAF-V600E achieved similar ORRs (50% (4/8) versus 43% (16/37); P = 1; Extended Data Fig. 4b), and the predictive accuracy of MSS/MSI status alone was 44% (sensitivity, 80%; specificity, 16%; PPV, 43%; NPV, 50%; Extended Data Fig. 4g).
We next classified tumor samples according to three molecular subtypes: MSS-RNF43wild-type, MSS-RNF43mutated and MSI-RNF43mutated. The ORR of the MSS-RNF43mutated subtype was significantly higher than those of the MSS-RNF43wild-type and MSI-RNF43mutated subtypes (73% versus 31% and 50%, respectively; P = 0.03) (Fig. 2a and Extended Data Fig. 4c). In agreement, accuracy for prediction of response combining MSS/MSI + RNF43 mutation status was 67% (sensitivity, 40%; specificity, 88%; PPV, 73%; NPV, 65%; Extended Data Fig. 4g).
Validation of MSS/MSI-RNF43 status and clinical response
To explore the generalizability of the results obtained in the discovery set, we sought to validate the predictive value of RNF43 mutations in an external independent cohort of patients treated with anti-BRAF/EGFR ± combinatorial therapies as second- or third-line therapy (n = 52). Overall, the ORR in the validation cohort was 27% (95% confidence interval (CI), 16.3–42%; 14/51 patients) with a median PFS (mPFS) of 4.4 months (95% CI, 4.1–5.9 months) and a median OS (mOS) of 8.5 months (95% CI, 6.9–14.2 months) (Supplementary Table 1). Consistent with the results in the discovery cohort, patients in the MSS-RNF43mutated subtype in the validation cohort also achieved a significatively higher ORR compared to those in the MSS-RNF43wild-type and MSI-RNF43mutated subtypes (54% versus 21% and 18%, respectively; P = 0.02; Fig. 2b and Extended Data Figs. 4d–f and 5a–d).
We next evaluated whether the observed increased ORR was a surrogate for improved outcomes measured by PFS and OS. The mPFS in the MSS-RNF43mutated subtype was 10.1 months (hazard ratio (HR), 0.36; 95% CI, 0.16–0.81), 4.1 months in the MSS-RNF43wild-type subtype and 4.4 months (HR, 0.74; 95% CI, 0.36–1.50) in the MSI-RNF43mutated subtype (Fig. 3a,b and Extended Data Fig. 6a,b). The MSS-RNF43mutated subtype also showed a trend toward better OS compared to the MSS-RNF43wild-type subtype (mOS of 13.6 versus 7 months; P = 0.07; HR, 0.46; 95% CI, 0.20–1.08; Fig. 3c,d and Extended Data Fig. 6c,d). In multivariate analysis, the MSS-RNF43mutated subtype maintained an independent association with OS (HR, 0.26; 95% CI, 0.10–0.71; P = 0.008) after adjusting for imbalance in prognostic factors such as age and metastatic site (Fig. 4a,b). Despite having a short PFS in response to anti-BRAF/EGFR therapies, patients with MSI-RNF43mutated status did not show a significantly lower mOS when compared to those with MSS-RNF43mutated status, most likely because of the positive impact of the treatment with immune-checkpoint inhibitors administered in six patients (12%) with MSI after progression on anti-BRAF/EGFR therapy (HR, 0.38; 95% CI, 0.09–1.48; P = 0.16; Fig. 4b).

a,b, Kaplan–Meier curves representing PFS of patients with RNF43wild-type (n = 29) and RNF43mutated (n = 23) tumors (a) and combined RNF43 and MSS/MSI status (MSS-RNF43wild-type (n = 28), MSS-RNF43mutated (n = 11) and MSI-RNF43mutated (n = 12)) (b). c,d, OS of patients with RNF43wild-type (n = 29) and RNF43mutated (n = 23) tumors (c) and combined RNF43 and MSS/MSI status (MSS-RNF43wild-type (n = 28), MSS-RNF43mutated (n = 11) and MSI-RNF43mutated (n = 12)) (d). Cox models were used to obtain HRs with 95% CIs, and the two-sided log-rank test was used for statistical comparisons without adjustment for multiplicity. Colors indicate molecular subtypes: RNF43wild-type tumors with or without MSS (yellow), RNF43mutated tumors with or without MSS (blue) and MSI-RNF43mutated tumors (gray). Significant values are shown in bold. m, months; NR, not reported; Ref., reference.

a,b, Analysis performed in the validation cohort (n = 52) for PFS (a) and OS (b). Univariate HRs along with 95% CIs are represented for each prognostic factor. P values were estimated by means of the two-sided log-rank test in the univariate analysis and by means of the Cox model in the multivariate analysis (two-sided). Significant values are shown in bold. ECOG, Eastern Cooperative Oncology Group performance; ICI, immune checkpoint inhibitor.
We also sought to explore whether, in addition to their power to predict response, the MSS/MSI-RNF43 molecular subtypes could anticipate refractoriness to anti-BRAF/EGFR ± combinatorial therapies. Of the 16 patients with refractory mCRCBRAF-V600E in the discovery and validation cohorts with PD as the best observed response, 13 (13/16, 81%) were MSS-RNF43wild-type, 3 (3/16, 19%) were MSI-RNF43mutated and none (0/16, 0%) were MSS-RNF43mutated. Strikingly, none of the 22 patients with MSS-RNF43mutated status in the two cohorts had PD as the best response (Extended Data Fig. 5d).
Predictive value of RNF43 mutations in MSS-mCRCBRAF-V600E
To further confirm the value of RNF43 mutations in predicting response to treatment in patients with MSS-mCRCBRAF-V600E tumors, we aggregated data from 68 sex- and age-matched patients treated in four different hospitals (Extended Data Fig. 7a–e). All patients received standard-of-care chemotherapies and antiangiogenic agents that did not involve anti-BRAF therapy. The median age at diagnosis was 60.5 years (range, 30–80 years); 68% of patients had right-sided tumors. With regard to MSS/MSI-RNF43 status, 22% of patients were MSS-RNF43mutated and 78% were MSS-RNF43wild-type, similar to the discovery and validation MSS tumor cohorts (29% and 71%, respectively) (Extended Data Fig. 7a–c and Supplementary Table 1). Moreover, the MSS discovery/validation and control cohorts presented with similar RNF43 mutation frequencies (29% (22/76) and 22% (15/68), respectively) (Extended Data Fig. 7b,c) as well as RNF43 mutation localization patterns (Extended Data Fig. 7d,e). Among 68 patients treated in the second- and third-line setting, those in the MSS-RNF43mutated subgroup achieved an mPFS of 1.3 months as compared to 1.6 months in the MSS-RNF43wild-type population (HR, 0.85; 95% CI, 0.40–1.79; P = 0.67). Similarly, we found no differences in mOS after the start of second-line therapy (5 months in patients with MSS-RNF43mutated tumors and 5.7 months in patients with MSS-RNF43wild-type tumors; HR, 0.93; 95% CI, 0.30–2.83; P = 0.89) (Fig. 5a–d). To corroborate these findings, we also evaluated mPFS in the first-line setting for the entire control cohort and found no significant differences in the MSS-RNF43mutated versus MSS-RNF43wild-type population (4.5 months versus 3.9 months, respectively; HR, 0.64; 95% CI, 0.36–1.15; P = 0.13) (Extended Data Fig. 8a–d). Altogether, these data suggest that RNF43 mutations have no prognostic relevance in patients with MSS-mCRCBRAF-V600E in our cohort and support their predictive value in response to anti-BRAF/EGFR combinatorial therapies in this clinical setting. Conversely, the predictive value of RNF43 mutations seen in MSS tumors was not observed in MSI tumors.

a–d, Kaplan–Meier curves representing PFS (a) and OS (b) for patients with mCRCBRAF-V600E undergoing anti-BRAF/EGFR therapy in the validation cohort (anti-BRAF/EGFR as second or third line, n = 39) and PFS (c) and OS (d) in patients in the control cohort not exposed to BRAF inhibition (n = 67 second and third treatment lines; Extended Data Fig.7). Cox models were used to obtain HRs with 95% CIs, and the two-sided log-rank test was used for statistical comparisons without adjustment for multiplicity. Colors refer to molecular subtypes: MSS-RNF43wild-type (yellow) and MSS-RNF43mutated (blue). Significant values are shown in bold.
Molecular profiles of RNF43 mutations in mCRCBRAF-V600E
To better understand the underlying molecular features of MSS-RNF43mutated CRCBRAF-V600E tumors, we next classified and characterized the observed RNF43 somatic mutations occurring in patients from both the discovery and validation cohorts. RNF43 mutations were identified in 21 of 22 (95%) MSI tumors and 22 of 76 (29%) MSS tumors. In addition to the different frequencies, the location and functional nature of the RNF43 mutations were fundamentally distinct in MSI and MSS tumors (Fig. 6a). All MSI RNF43-mutated tumors carried a hotspot mutation encoding p.G659fs*41 at the C-terminal, which is caused by a DNA slippage error at the seven-guanine repeat region, typical in MSI tumors lacking full DNA repair machinery, and classified as moderate loss of function with retained wild-type-like functional properties26. Conversely, the mutations occurring in MSS tumors were not recurrent and spanned the N-terminal domain of the protein, including the extracellular (EC), protease-associated (PA), transmembrane (TM), RING finger (RING) and DVL2-binding (DVL) domains. We identified 7 missense mutations in these domains and 16 that led to protein truncations predicted to cause loss of function27,28 (Fig. 6a,b). To validate the biological impact of the RNF43 missense mutations detected in the MSS-mCRCBRAF-V600E tumors of our patients, we performed luciferase reporter assays27,28. Compared to wild-type RNF43, six of seven (86%) of the RNF43 mutations analyzed (encoding I48T, A73V, A169T, H292Y, R296H and W302R substitutions) behaved as loss-of-function variants (Fig. 6c,d). The corresponding inactivated RNF43 mutants lost the capacity to ubiquitinate and degrade FZD/WNT receptors, resulting in an accumulation of these receptors on the cell membrane and in high WNT signal levels in the luciferase reporter assays (Fig. 6c–e). Because of this effect, RNF43 loss-of-function mutations are also referred to as ‘WNT-hyperactivating’ mutations26,27,28.

a, Distinct localization and functional impact of RNF43 mutations among patients with mCRCBRAF-V600E treated with anti-BRAF/EGFR therapy according to MSS/MSI and RNF43 molecular status (n = 98). MSI CRCs (n = 21) carried a mutation encoding G659fs, while MSS CRCs were divided into MSS-RNF43mutated (n = 22) and MSS-RNF43wild-type (n = 54) tumors. Numbers indicate the amino acid residue in the RNF43 protein sequence. The MSS-RNF43mutated subtype harbored mutations mainly in the RNF43 N-terminal domain. Colors indicate the effect of the mutation on protein function: loss of function (LOF) (red), moderate loss of function (yellow) and normal function (blue); symbols reflect MSS/MSI status (circle and triangle, respectively) and the presence of a compound mutation (discontinuous border). b, Illustration of the ‘cross-brace’ topology of the RING domain containing a highly conserved sequence of cysteine–histidine residues that coordinates two atoms of zinc and four hydrophobic residues that are involved in binding to E2 (canonical sequence, CX2CX(9–39)CX(1–3)HX(2–3)CX2CX(4–48)CX2C). Figure adapted from ref. 36, Frontiers Media (CC BY 4.0 license). Mutations encoding H292Y, R296H and W302R in the RING domain of RNF43 are circled in red, while the mutation encoding M313R is located right outside the RING protein domain and likely for this reason did not affect RNF43’s ubiquitinase function. The four conserved residues are shown in blue. c, Western blot quantification of Flag-tagged RNF43 protein ectopically expressed in the HEK293T cell line; β-actin, loading control. This experiment was repeated twice obtaining the same protein expression levels. d, In vitro luciferase reporter assays representing levels of β-catenin activation (y axis) upon ectopic expression of RNF43 mutants (x axis) in HEK293T cells, following stimulation with Wnt3A conditioned medium (CM). One representative experiment is shown. The assay was performed three times with basically identical results. The statistical significance of all mutants relative to wild-type protein was obtained using a two-sided Student’s t-test (**P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant). Empty vector (EV) and EV + WNT (control conditions) are shown in gray, RNF43 alterations that behaved as loss-of-function variants are shown in red and wild-type protein and the M313R variant are shown in blue. e, Graphical representation of the impact of RNF43 mutations (1) in impairing the ubiquitinase activity of the protein (2), resulting in the accumulation of FZD/WNT receptors in the cell membrane (3). Ub, ubiquitin.
Mutations detected in other genes of the WNT pathway, such as APC and CTNNB1, are shown in Supplementary Table 1 and did not correlate with ORR (Extended Data Fig. 9a,b). Moreover, no correlation was observed of β-catenin protein expression levels and localization with response to anti-BRAF/EGFR ± combinatorial therapies, MSS/MSI status or RNF43 mutation status in 33 mCRCBRAF-V600E tumors from the discovery cohort (Extended Data Fig. 9c,d).
Discussion
mCRCBRAF-V600E represents an entity of its own with particular phenotypic features and with crucial implications in terms of prognosis2,3. Hence, there is a critical need for new clinical–biological insights that can lead to therapeutic improvements and the identification of new biomarkers of response. However, identification of biomarkers in this patient population, as well as development of new therapeutic strategies, has been challenging in this particular tumor type, given its underrepresentation in randomized clinical trials due to its low frequency (up to 12% of mCRC) and the high tumor burden and poor clinical conditions that impair inclusion of these patients in clinical trials29. Indeed, some of the current recommendations for standard-of-care treatments are based on subgroup analyses of phase 3 clinical trials3,30,31. On the other hand, despite the meaningful clinical activity observed in clinical trials evaluating BRAF inhibitor-based combinations, not all patients respond the same, and some responses are relatively short. This disparity in terms of treatment benefit highlights the biological heterogeneity of mCRCBRAF-V600E and justifies better molecular characterization to optimize treatment outcomes.
Through pursuing an unbiased genomic analysis of responders versus nonresponders, we detected a strong signal in the RNF43 locus in association with clinical outcome in response to anti-BRAF/EGFR-based therapies that was confirmed in a validation cohort (Fig. 3a–d) but not observed in a sex- and age-matched control cohort of patients receiving standard chemotherapy ± antiangiogenic agents (Fig. 5a–d and Extended Data Fig. 8a–d). Our study uncovers a previously unknown value of RNF43 mutations, occurring in 29% of MSS-mCRCBRAF-V600E tumors, in predicting response and clinical outcome. In contrast to patients with MSI mCRC, the patient population with MSS-mCRCBRAF-V600E currently lacks biomarkers to guide treatment decision-making. Although previous analyses had associated tumor-based transcription subtypes with response32, our study reports a potential genomic prediction biomarker to anti-BRAF/EGFR-based combinations in the patient population with mCRCBRAF-V600E. Specifically, we found that within the MSS group, which represents 70% of patients with mCRCBRAF-V600E, the occurrence of a RNF43 mutation was associated with improved ORR and, importantly, longer PFS and OS with anti-BRAF/EGFR strategies (Figs. 2a,b, 3a–d and 5a–d and Extended Data Figs. 4a–g and 5a–f).
Overall, this finding has two main implications. First, incorporating RNF43 mutation as a routine biomarker could contribute to defining the optimal treatment sequence in patients with MSS-mCRCBRAF-V600E according to their predicted response profile. Second, it uncovers a cross-talk between MAPK and RNF43–WNT pathways in the antitumor activity of BRAF/EGFR-targeting therapy, which might be exploited as a future potential therapeutic target.
The mechanistic basis underlying the clinical success of concomitant BRAF/EGFR inhibition builds upon preclinical evidence of a rapid feedback compensation for EGFR mediated by the activation of CRAF or by the transactivation of CRAF–BRAF heterodimers13,14. Our findings underscoring the potential role of RNF43 in modulating response to anti-BRAF/EGFR therapy point to an interplay between the MAPK and WNT signaling pathways in MSS-mCRCBRAF-V600E tumors. Specifically, our in vitro experiments show that most RNF43 mutations detected in MSS tumors of responding patients have a loss-of-function effect, which contrasts with RNF43 mutations described in MSI tumors, where the protein retains its function26. In our patient datasets, loss of RNF43 would impair degradation of WNT/FZD receptors, leading to activation of the WNT pathway. While the molecular intricacies through which the MAPK and WNT pathways cross-talk to modulate the antitumor activity of this treatment combination need to be studied in detail, our clinical findings are consistent with preclinical studies describing a role for WNT activation in antagonizing MAPK-driven proliferation to preserve an equilibrium with differentiation of intestinal stem cells33. It is therefore plausible to speculate that a similar mechanism of RNF43 loss-dependent WNT activation may be restraining MAPK signaling in MSS mCRC tumors and synergizing with pharmacological blockade of the pathway. Of note, we found no correlation of response with the presence of APC or CTNNB1 mutations or β-catenin protein expression or localization in mCRCBRAF-V600E tumors (Extended Data Fig. 9a–d and Supplementary Table 1), suggesting that noncanonical WNT pathways34,35, rather than canonical (β-catenin-dependent) signaling, might be involved in modulation of anti-BRAF/EGFR activity.
Despite our study representing, to our knowledge, the largest genetic biomarker analysis to date on this patient population, the overall number of patients remains limited owing to the rarity of BRAFV600E mutation in mCRC and the difficulties in accessing clinically annotated cohorts with comprehensive genomic profiling. Furthermore, while our genomic analyses uncover a robust association of RNF43 mutations with clinical outcome, the inherent heterogeneity of our real-world patient cohorts receiving treatment in four different hospitals and undergoing molecular profiling with different multigene panels represents another limitation to be addressed in prospective, standardized biomarker studies. The lack of randomized data represents a constraint for analysis of the predictive versus prognostic role of RNF43 mutations. In this regard, our efforts to identify control patients exposed to chemotherapies and antiangiogenic agents during the same time period and at the same hospitals that contributed data for the discovery and validation cohorts are the best mitigation available, also taking into account that similar NGS platforms were used for molecular stratification. Finally, despite our in vitro analysis supporting a loss-of-function effect of most RNF43 mutations detected in MSS tumors from responding patients, the mechanistic basis for how altered WNT signaling modulates MAPK pathway activation in response to anti-BRAF/EGFR agents remains to be fully elucidated.
In summary, RNF43 mutations represent a new biomarker that warrants further validation for its potential to help prioritize anti-EGFR/BRAF combinations in selected patients with mCRCBRAF-V600E who are more likely to derive benefit and identify those patients for whom alternative treatment approaches are needed. Future research should explore incorporating this biomarker in routine testing along with BRAF and MSS/MSI status and evaluate their integration with other transcriptomic, microbiome or microenvironmental indicators for optimizing the clinical management of this heterogeneous and complex disease.
Methods
Predictive and prognostic value analysis
To assess the predictive value of RNF43 status, we aggregated the clinical and genetic data of 68 patients from four different cohorts (hospitals participating in the present work) treated with standard-of-care regimens only (no exposure to anti-BRAF therapies) whose tumors harbored BRAFV600E mutation and with information on the status of MSI and RNF43 mutations. These patients received in total 135 chemotherapy regimens with or without antiangiogenic drugs during the first, second or third line of therapy. To control for potential confounding factors, we excluded (1) treatments in the first line and (2) patients whose tumors were MSI and who received anti-PD1/PD-L1 therapies during the disease course. A total of 67 treatment lines (second and third line) in patients with MSS were analyzed. To compare the prognosis of patients with MSS-RNF43mutated and MSS-RNF43wild-type tumors, PFS and OS endpoints were estimated using the Kaplan–Meier method. Mixed-effects Cox models considering patient ID as a random effect, to adjust for the intra-patient variability in patients with more than one line of therapy, were used to obtain HRs with 95% CIs (Supplementary Table 1).
Ethics committee approval
The study was approved by each investigational site’s institutional review board/ethics committee: Vall d’Hebron Institute of Oncology (VHIO), Barcelona, Spain; Veneto Institute of Oncology IRCCS, Padova, Italy; Azienda Ospedaliero-Universitaria Pisana, University of Pisa, Pisa, Italy; and Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy. The research was conducted in accordance with the Declaration of Helsinki and local data protection laws. All patients were provided with written informed consent before enrollment. All data provided are anonymized in line with applicable laws and regulations.
Radiological response evaluation
All cases were reviewed by board-certified radiologists with experience in oncology imaging and clinical trials. Radiological response for each patient was classified following the principles of RECIST 1.1 (ref. 22): CR (disappearance of the lesion or reduction in the short axis to <10 mm in the case of a pathological lymph node), PR (a decrease by at least 30% in the long axis for visceral or soft tissue disease and the short axis for pathological lymph nodes), PD (an increase of at least 20% in the long axis in the case of visceral or soft tissue disease and the short axis in the case of pathological lymph nodes) or SD (when the lesion did not fulfill the criteria for PR or PD). As is commonly the case in clinical practice for aggressive tumors, some patients included in the study had no image available due to evident progression as per rapid clinical deterioration, increase in the levels of tumor markers in plasma (CEA) and progression of nontarget disease. These patients were considered to have clinical PD.
DNA extraction
DNA extraction from tumor samples was performed with the automated system Maxwell 16 FFPE plus LEV DNA purification kit (Promega), and quality and concentration were measured with a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). cfDNA was extracted from 1 ml of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen), based on the manufacturer’s recommendations, and quantified using the highly sensitive kit for the Qubit, dsDNA HS (High Sensitivity) Assay Kit (Thermo Fisher Scientific).
WES
WES was performed in 55 baseline biological samples available at VHIO’s sample biobank (19 germline DNA, 22 tumor DNA, 5 patient-derived xenograft DNA and 9 plasma cfDNA) from 28 patients from the discovery cohort. Genomic libraries were prepared using 10–15 ng of cfDNA and the ThruPLEX DNA or Plasma-seq Kit (Rubicon Genomics; now commercialized as the SMARTer ThruPLEX plasma-seq kit by Takara Bio)37,38. Quality control of libraries was carried out in TapeStation (Agilent), and amplified profiles of ~300 base pairs were considered for downstream analysis. Capture of the genomic coding regions for WES was performed using the SureSelect Human All Exon V5 or V6 kit (Agilent), with a target sequencing output of 12 Gb (100×) for genomic DNA and 36 Gb (300×) for tumor and cfDNA. Sequencing was carried out on a HiSeq or NovaSeq Illumina sequencing platform.
NGS cancer gene panel
Tumor samples from the discovery cohort that did not have WES data (n = 18) and from the validation (n = 52) and control (n = 68) cohorts were genetically analyzed using a VHIO in-house NGS test of 430 cancer genes or a Foundation Medicine or Caris Life Sciences commercial NGS platform39.
Bioinformatics
Data processing and analysis (WES)
The WES samples (FASTQ files) were processed using sarek (v2.7.1)40. Briefly, the following steps were performed: quality filtering and adaptor trimming with TrimGalore; alignment to the reference genome with BWA; marking of duplicates with GATK4 MarkDuplicates; recalibration of scores with GATK4 BaseRecalibrator and ApplyBSQR; and computing of pileups with SAMtools. Variant calling was performed using four different tools: Mutect2, Strelka2, MSIsensor and Control-FREEC (tumor-only and pair mode). Filtering of the variants generated by Mutect2 was conducted using GATK4 GetPileupSummaries, CalculateContamination and FilterMutectCalls. Annotation of the variants was performed using snpEff. MultipleQC and statistics were generated using FastQC, Qualimap, SAMtools, VCFtools and MultiQC. We used a public Panel of Normals (PON) obtained from the public repository of GATK at gs://gatk-best-practices/somatic-hg38/1000g_pon.hg38.vcf.gz and a BED file containing all the genomic intervals of the kits used to generate the WES samples. Mutect2 variants corresponding to the tumor-only samples were refiltered with GATK4 FilterMutectCalls using a value of 10 for the parameter ‘-max-events-in-region’. These variants were subsequently reannotated with snpEff with the same genome reference, version and settings that were used in the processing pipeline. The generated mutational data were subsequently summarized, postprocessed, filtered and validated for downstream analysis.
Mutational enrichment analysis
Two different analyses were performed on the discovery cohort to determine which genes were enriched for somatic mutations in the responder and nonresponder groups. In the first analysis, we summarized the number of mutations, accumulated mutated allele frequency and number of samples/patients per gene. In the second analysis, we computed somatic enrichment P values and q values for each group using dNdScv25 (GRCh38). This tool models the background mutation rate of each gene by combining local information (synonymous mutations in the gene) and global information (nonsynonymous mutations in the gene and other covariates) and controlling for the sequence composition of the gene and mutational signatures25.
Functional classification of RNF43 mutations
Functional classification derived from previous in vitro and in vivo studies
To initially assess the potential biological consequences of the identified RNF43 mutations, we applied a recently developed functional classification derived from in vitro and in vivo studies26,27,28. Accordingly, RNF43 mutations can be classified as (1) wild-type like, (2) activators of the WNT–β-catenin pathway or (3) hyperactivators of the WNT–β-catenin pathway26,27,28. Mechanistically, wild-type-like mutants maintain their capability to repress the WNT–β-catenin pathway and activators lose their ubiquitinase function, leading to an increased abundance of FZD/WNT receptors in the cellular membrane and activation of the WNT–β-catenin pathway, while hyperactivators exert a dominant-negative effect on the wild-type protein, completely blocking ubiquitination of FZD/WNT receptors and thereby leading to extremely high levels of WNT–β-catenin pathway activation26,27,28.
Luciferase reporter assays with RNF43 mutation expression vectors
To further demonstrate the biological impact of the identified RNF43 missense mutations, we performed luciferase reporter assays with ectopic expression of the RNF43 mutants. Flag-tagged RNF43 mutation expression plasmids were generated using the New England Biolabs Q5 Mutagenesis Kit27,28. All plasmids were full-length sequence verified.
HEK293T cells were used for the assays, cultured in DMEM (Gibco) supplemented with 10% (vol/vol) FBS (Gibco). Cells were cultured in a humidified incubator maintained at 37 °C with 5% CO2. Cells tested negative for mycoplasma based on the real-time PCR method at Eurofins GATC-Biotech (Konstanz, Germany). Identity of the HEK293T cells was confirmed by the Erasmus Molecular Diagnostics Department, using PowerPlex-16 STR genotyping (Promega). For the β-catenin reporter assays, HEK293T cells were seeded in 24-well plates and transfected with 100 ng WRE Wnt/β-catenin reporter, 100 ng RNF43 expression plasmid (wild-type RNF43, mutant RNF43 or empty vector) and 10 ng CMV-Renilla expression plasmid, using Lipofectamine 2000 (Thermo Fisher Scientific) as transfection reagent. L-Wnt3A or L-control conditioned medium (diluted 30-fold in normal growth medium) was added after 16 h. Cells were lysed in passive lysis buffer (Promega) 48 h after transfection. Next, the firefly and Renilla luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega) using a LumiStar Optima luminescence counter (BMG LabTech). β-catenin reporter activity was measured in triplicate. All β-catenin reporter values were normalized to the value obtained for cells with empty vector exposed to L-control conditioned medium, which was arbitrarily set to 1. One representative experiment is shown. The assay was performed three times with basically identical results. Proper expression of Flag-tagged RNF43 was analyzed using fluorescent western blotting as described below.
Immunoblotting analysis
Immunoblotting was carried out using standard methods. HEK293T cells transfected with RNF43 were lysed in 2× Laemmli sample buffer with 0.1 M dithiothreitol and heated for 10 min at 95 °C. Proteins were separated by 10% SDS–PAGE and then transferred to an Immobilon-P PVDF membrane (MilliporeSigma). The membrane was blocked for 1 h with Odyssey Blocking Buffer (LI-COR Biosciences) at room temperature and incubated overnight with primary antibodies at 4 °C. After washing three times with 0.05% Tween-20 in TBS (TBST) buffer for 10 min, the membrane was incubated for 1 h with IRDye 680 Goat Anti-Mouse (1:10,000; LI 926-68070, LI-COR Biosciences) and then washed three times with TBST for 10 min. The membrane was scanned on the Odyssey Infrared Imaging System (LI-COR Biosciences). Primary antibodies used were mouse anti-FLAG (1:1,000; F1804, Sigma-Aldrich) and mouse anti-β-actin (1:1,000; sc-47778, Santa Cruz).
Immunostaining of β-catenin in CRCBRAF-V600E tumors
Immunohistochemical analysis was performed using a primary antibody against human β-catenin (prediluted, Beta-Catenin Mouse Monoclonal Antibody, clone 14, 760-4242, Cell Marque) in Benchmark ULTRA (Ventana Medical Systems). Labeling was performed using ULTRA Cell Conditioning 1 (ULTRACC1) (950-224, Roche) antigen retrieval buffer for 64 min at 95 °C, followed by incubation with primary antibody for 32 min at 36 °C and detection with the Ventana UltraView Universal DAB Detection Kit (760-500, Roche). Immunohistochemistry was scored semiquantitatively by one pathologist (R.F.) using an H-score (for the cytoplasm, membrane and nucleus separately). H-score was obtained by multiplying the proportion of cells showing cytoplasmic, membrane or nuclear staining and the intensity of staining (0, no staining; 1, weak; 2, moderate; 3, strong).
Statistical analysis
A descriptive analysis of all included variables in the study was performed. Continuous variables were expressed as the median and interquartile range, and categorical variables were expressed as absolute values and percentages. ORR was estimated in all subgroups based on RECIST version 1.1 criteria along with 95% CIs, and Fisher’s exact test was used to assess statistical significance. Accuracy, sensitivity, specificity, and PPV and NPV were calculated to quantify the diagnostic performance of each potential biomarker.
PFS was defined as the time from anti-BRAF/EGFR therapy initiation to disease progression or death, whichever occurred first. OS was defined as the time from anti-BRAF/EGFR therapy initiation to death from any cause. PFS and OS were estimated using the Kaplan–Meier method and compared by the log-rank test. Cox proportional-hazard models were used to obtain HRs with 95% CIs. Formal statistical testing was only used in the validation cohort. No data imputation was performed. All tests were two sided with a value of P < 0.05 considered statistically significant without adjustment for multiple comparisons. All statistical analyses were performed using R statistical software.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Reference genome GRCh38 was used for alignment. The FASTQ files corresponding to the WES data from clinical samples analyzed in the paper have been deposited in the European Genome-phenome Archive (EGA, https://ega-archive.org) under study ID EGAS00001006247 and dataset ID EGAD00001008755 with appropriate measures for controlled access (DUO: 0000020, DUO: 0000021). Further information about EGA can be found on https://ega-archive.org, “The European Genome-phenome Archive in 2021” (https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab1059/6430505). Clinical and genetic data for the patients are available in Supplementary Table 1. Source data are provided with this paper.
Code availability
The code for the pipeline that was used to process all samples can be found at https://github.com/nf-core/sarek. Sarek is a Nextflow-based pipeline that integrates all the processing, mapping, variant calling and quality control steps25. The code used for postprocessing, filtering, validation and analysis of the mutational data is available at https://github.com/jfnavarro/scitron.
References
Di Nicolantonio, F. et al. Precision oncology in metastatic colorectal cancer—from biology to medicine. Nat. Rev. Clin. Oncol. 18, 506–525 (2021).
Morris, V. et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin. Colorectal Cancer 13, 164–171 (2014).
Cremolini, C. et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 16, 1306–1315 (2015).
Yaeger, R. et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33, 125–136 (2018).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304 (2018).
Koo, B. K. et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669 (2012).
Nusse, R. & Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Hyman, D. M. et al. Vemurafenib in multiple nonmelanoma cancers with BRAFV600 mutations. N. Engl. J. Med. 373, 726–736 (2015).
Flaherty, K. T. et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010).
Sosman, J. A. et al. Survival in BRAFV600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 366, 707–714 (2012).
Kopetz, S. et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J. Clin. Oncol. 28, 3534 (2010).
Kopetz, S. et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J. Clin. Oncol. 33, 4032–4038 (2015).
Prahallad, A. et al. Unresponsiveness of colon cancer to BRAFV600E inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012).
Corcoran, R. B. et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235 (2012).
van Geel, R. et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 7, 610–619 (2017).
Kopetz, S. et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J. Clin. Oncol. 39, 285–294 (2021).
Ahronian, L. G. et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 5, 358–367 (2015).
Kopetz, S. et al. Encorafenib, binimetinib, and cetuximab in BRAFV600E-mutated colorectal cancer. N. Engl. J. Med. 381, 1632–1643 (2019).
Van Cutsem, E. et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAFV600E-mutant metastatic colorectal cancer: safety lead-in results from the phase III BEACON colorectal cancer study. J. Clin. Oncol. 37, 1460–1469 (2019).
Tabernero, J. et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAFV600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J. Clin. Oncol. 39, 273–284 (2021).
Kopetz, S. et al. Molecular correlates of clinical benefit in previously treated patients (pts) with BRAFV600E-mutant metastatic colorectal cancer (mCRC) from the BEACON study. J. Clin. Oncol. 39, 3513 (2021).
Schwartz, L. H. et al. RECIST 1.1—standardisation and disease-specific adaptations: perspectives from the RECIST Working Group. Eur. J. Cancer 62, 138–145 (2016).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Martincorena, I. et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041 (2017).
Tu, J. et al. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci. Rep. 9, 18557 (2019).
Yu, J. et al. The functional landscape of patient-derived RNF43 mutations predicts sensitivity to Wnt inhibition. Cancer Res. 80, 5619–5632 (2020).
Li, S. et al. Commonly observed RNF43 mutations retain functionality in attenuating Wnt/β-catenin signaling and unlikely confer Wnt-dependency onto colorectal cancers. Oncogene 39, 3458–3472 (2020).
Van Cutsem, E., Cervantes, A., Nordlinger, B. & Arnold, D. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 25, iii1–iii9 (2014).
Van Cutsem, E. et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 30, 3499–3506 (2012).
Tabernero, J. et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 16, 499–508 (2015).
Middleton, G. et al. BRAF-mutant transcriptional subtypes predict outcome of combined BRAF, MEK, and EGFR blockade with dabrafenib, trametinib, and panitumumab in patients with colorectal cancer. Clin. Cancer Res. 26, 2466–2476 (2020).
Kabiri, Z. et al. Wnt signaling suppresses MAPK-driven proliferation of intestinal stem cells. J. Clin. Invest. 128, 3806–3812 (2018).
Tsukiyama, T. et al. A phospho-switch controls RNF43-mediated degradation of Wnt receptors to suppress tumorigenesis. Nat. Commun. 11, 4586 (2020).
Radaszkiewicz, T. et al. RNF43 inhibits WNT5A-driven signaling and suppresses melanoma invasion and resistance to the targeted therapy. eLife 10, e65759 (2021).
Garcia-Barcena, C., Osinalde, N., Ramirez, J. & Mayor, U. How to inactivate human ubiquitin E3 ligases by mutation. Front. Cell Dev. Biol. 8, 39 (2020).
Toledo, R. A. et al. Exome sequencing of plasma DNA portrays the mutation landscape of colorectal cancer and discovers mutated VEGFR2 receptors as modulators of antiangiogenic therapies. Clin. Cancer Res. 24, 3550–3559 (2018).
Hastings, R. K. et al. Longitudinal whole-exome sequencing of cell-free DNA for tracking the co-evolutionary tumor and immune evasion dynamics: longitudinal data from a single patient. Ann. Oncol. 32, 681–684 (2021).
Randon, G. et al. Tumour mutational burden predicts resistance to EGFR/BRAF blockade in BRAF-mutated microsatellite stable metastatic colorectal cancer. Eur. J. Cancer 161, 90–98 (2022).
Garcia, M. et al. Sarek: a portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Res 9, 63 (2020).
Acknowledgements
VHIO would like to acknowledge the Cellex Foundation for providing research facilities and equipment, the FERO Foundation for their funding support, the Consorcio Centro de Investigación Biomédica en Red de Cáncer (CIBERONC, CB16/12/00259) from the Institute of Health Carlos III (ISCIII), Ministry of Science and Innovation, and the Department of Health (Generalitat de Catalunya, SLT008/18/00198 and SLT008/18/00205) for their support on this research. Authors acknowledge financial support from the State Agency for Research (Agencia Estatal de Investigación) (CEX2020-001024-S / AEI / 10.13039 /501100011033). This research is funded by the SCITRON program; Novartis funded the genomics characterization by WES of samples from 28 patients from the discovery cohort and had no influence on data analysis/interpretation or writing of the paper (3003145512 to R.A.T.). S.Li. is financially supported by a Chinese Scholarship Council PhD fellowship (201909370083 to S. Li). R.P.-L. is supported by a CRIS Foundation Talent Award (TALENT19-05), the FERO Foundation, the Instituto de Salud Carlos III-Investigación en Salud (PI18/01395 and PI21/01019 to R.P.-L.) and the Prostate Cancer Foundation (Young Investigator Award). This work was supported by the Miguel Servet-I Research Award from ISCIII of the Ministry of Economy (CP17/00199 to R.A.T.), the Olga Torres Foundation Award to emerging researchers (2601 to R.A.T.), the ISCIII-FEDER (PI17/00947 and PI20/00968 to E.E.), and the Fundación AECC (CLSEN19001ELEZ to E.E.) and Ministry of Science and Innovation (Europa Redes y Gestores, ECT2020-000827 to E.E.).
Author information
Authors and Affiliations
Contributions
E.E., J.T. and R.A.T. conceived and designed the study. E.E., J.R., A.B.M.-C., C.A., A.G., P.N., R.I., A.M., M.M.G., G.R., C.C., S. Lonardi, F.P. and R.A.T. were involved in sample collection and data acquisition. E.E., J.R., J.F., A.B.M.-C., C.A., S. Li, R.F., A.V., R.S., C.C., S. Lonardi, F.P., R.D., J.T. and R.A.T. performed experiments, analysis and data interpretation. J.G.-R., A.P., D.P.K. and D.G. were involved in the project and study management. G.V., R.C. and R.D. performed the data and statistical analyses. K.B. and R.P.-L. performed the imaging and radiological analyses. A.B.M.-C. and A.V. performed genomic analyses. J.F. performed the bioinformatics analyses of the WES data and other computational analyses. G.K. performed computational analysis. C.A. and R.A.T. created Figs. 1 and 6e and Extended Data Figs. 1a, 2b and 3a with BioRender.com. R.A.T. wrote the manuscript, and E.E., J.R., R.S., A.B.M.-C., C.A., C.C., S. Lonardi, F.P., R.D. and J.T. participated in the review and editing. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
C.C. reports receiving honoraria and speaker’s bureau compensation from Servier, Bayer, Merck, Amgen, Pierre Fabre, MSD, Roche and Nordic Pharma and research grants from Merck, Servier and Amgen. D.G. was an employee of Novartis and was a shareholder while this work was being conducted. D.P.K. is an employee of Novartis. E.E. reports receiving honoraria for an advisory role, travel grants and research grants (past 5 years) from Hoffmann-La Roche, Bristol-Myers Squibb, Servier, Amgen, Merck Serono, Array Biopharma, Sanofi and Bayer. Her institution received honoraria due to her investigator contribution in clinical trials from Array Biopharma, MSD, Abbvie, Amgen, GlaxoSmithKline, AstraZeneca, Merck Sharp & Dohme, Bristol-Myers Squibb, Novartis, Boehringer Ingelheim, Hoffmann-La Roche, Medimmune, Pierre-Fabre and Sanofi Aventis. F.P. received honoraria from Amgen, Merck Serono, Lilly, Sanofi, Bayer, Servier, MSD, AstraZeneca and Organon and research grants from AstraZeneca and BMS. G.K. is an employee of Novartis. G.V. has received a speaker’s fee from MSD and Pierre Fabrer and has held an advisory role with AstraZeneca. J.R. declares receiving honoraria from Sanofi and travel and accommodation expenses from Amgen, Merck and Sanofi. J.T. reports having a personal financial interest in the form of a scientific consultancy role for Array Biopharma, AstraZeneca, Avvinity, Bayer, Boehringer Ingelheim, Chugai, Daiichi Sankyo, F. Hoffmann-La Roche, Genentech, HalioDX, Hutchison MediPharma International, Ikena Oncology, Inspirna, IQVIA, Lilly, Menarini, Merck Serono, Merus, MSD, Mirati, Neophore, Novartis, Ona Therapeutics, Orion Biotechnology, Peptomyc, Pfizer, Pierre Fabre, Samsung Bioepis, Sanofi, Seattle Genetics, Scandion Oncology, Servier, Sotio Biotech, Taiho, Tessa Therapeutics and TheraMyc, as well as educational collaboration with Imedex, Medscape Education, MJH Life Sciences, PeerView Institute for Medical Education and Physicians Education Resource (PER). P.N. reports receiving honoraria or consultation fees from Novartis, Bayer and MSD Oncology and had travel and accommodation paid for or reimbursed by Novartis. R.A.T. reports receiving a research grant related to this study from Novartis and research grants unrelated to this study from AstraZeneca and Beigene. R.D. declares an advisory role for Roche and Boehringer Ingelheim and received a speaker’s fee from Roche, Boehringer Ingelheim, Ipsen, Amgen, Servier, Sanofi, Libbs and Merck Sharp & Dohme and research grants from Merck and Pierre Fabre. R.P.-L. has acted in a consulting or advisory role for Roche, and she has participated in speaker bureaus sponsored by Roche and Pfizer. She is also the principal investigator in research grants to VHIO funded by AstraZeneca and Roche. S. Lonardi declares no conflict of interest inside the scope of the present work but declares interests in other projects from a consulting or advisory role (Amgen, Merck Serono, Lilly, AstraZeneca, Incyte, Daiichi-Sankyo, Bristol-Myers Squibb, Servier and MSD); speaker’s bureau compensation from Roche, Lilly, Bristol-Myers Squibb, Servier, Merck Serono, Pierre-Fabre, GSK and Amgen; and research funding from Amgen, Merck Serono, Bayer, Roche, Lilly, AstraZeneca and Bristol-Myers Squibb. The other authors declare no conflicts of interest.
Peer review
Peer review information
Nature Medicine thanks Jan Paul Medema, Fortunato Ciardiello and Dominik Modest for their contribution to the peer review of this work. Primary Handling Editor: Joao Monteiro, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Patients’ characteristics.
A) Patient and therapy flowchart. Specific clinical and genetic characteristics of mCRCBRAF-V600E compared to mCRCBRAF-wild-type tumors. Schematic description of the anti-BRAF/EGFR combinatorial therapies (doublet vs triplet) for patients with mCRCBRAF-V600E. B) Clinical characteristics of patients in the discovery (n = 46) and validation (n = 52) cohorts. Abbreviations: ECOG PS, Eastern Cooperative Oncology Group Performance Status; ICIs, immune checkpoint inhibitors; MSI-high, microsatellite instability high; MSS, microsatellite stable.
Extended Data Fig. 2 Bioinformatics pipeline for genomic data analysis.
A) Description of the Sarek bioinformatics pipeline, a Nextflow-based pipeline that integrates all the processing, mapping, variant calling, and QC steps40, used for the WES analysis. B) WES based on responders vs non-responders from the discovery cohort (n = 28) followed by dNdScv25 maximum-likelihood unbiased mutation enrichment analysis was used as the genomic biomarker discovery strategy on 55 biological samples from mCRCBRAF-V600E patients collected at baseline to anti-BRAF/EGFR ± MEKi therapies. The FastQ files have been deposited in the European Genome-phenome Archive (EGA, https://ega-archive.org) biorepository. The code of the bioinformatics pipelines can be found at https://github.com/nf-core/sarek and at https://github.com/jfnavarro/scitron. Abbreviations: cfDNA, cell-free DNA; CNA, copy number alteration; QC, quality control; PDX, patient-derived xenograft.
Extended Data Fig. 3 The utility of liquid biopsy to assess BRAF and RNF43 mutational status in patiets with mCRCBRAF-V600E treated with anti-BRAF/EGFR combinatorial therapies.
A) WES was performed in 55 baseline biological samples from 19 gDNA, 22 tDNA, 9 cfDNA, and 5 pdxDNA from 28 patients with mCRCBRAF-V600E from the discovery cohort treated with anti-BRAF/EGFR combinatorial therapies. The presence of the BRAFV600E and RNF43 mutations was interrogated in tumor-derived and plasma-derived samples. B) Mutation allele frequencies (MAF) of the BRAF and RNF43 specific mutations are shown to gDNA, tDNA, and pdxDNA from patients with multiple samples which are available. C) BRAFV600E and RNF43 mutations were detected not only in tDNA and/or pdxDNA but also in available cfDNA samples, suggesting the utility of liquid biopsy for the assessment of these biomarkers. Abbreviations: gDNA, germline DNA; tDNA, tumor DNA; cfDNA, cell-free DNA; MSI, microsatellite instability high; MSS, microsatellite stable; pdxDNA, patient-derived xenograft DNA.
Extended Data Fig. 4 Overall response rate (ORR) (%) of the discovery (n = 45, A–C) and validation (n = 50, D–F) cohorts and diagnostic performance analyses (G) defined by MSS/MSI and RNF43 mutation statuses.
A) Patients with RNF43mutated tumors exhibited an increased ORR (63%) compared to patients with RNF43wild-type (31%). B) There were no significant differences between ORRs according to MSI/MSS status (50% and 43%, respectively). C) Interestingly, patients with mCRCBRAF-V600E with MSS-RNF43mutated tumors demonstrated increased ORR (73%) compared to the other groups, as MSI-RNF43mutated and MSS-RNF43wild-type (50% and 31%, respectively) (P = 0.03). Chi-square test was used for the statistical analysis. D–F) ORR of the validation cohort including 50 patients with mCRCBRAF-V600E tumors treated with anti-BRAF/EGFR combinatorial therapies in 2nd or 3rd line. D) RNF43mutated group exhibited an increased ORR (36%) compared to RNF43wild-type group (21%). E) There were no significant differences between MSI/MSS status (31% and 17%, respectively). F) Interestingly, MSS-RNF43mutated group showed increased ORR (54%) compared to the other groups, MSI-RNF43mutated and MSS-RNF43wild-type (18% and 21%), respectively (P = 0.02). Chi-square test was used for the statistical analysis (*P < 0.05). G) Table showing results of the accuracy, sensitivity, specificity, and positive (PPV) and negative predictive value (NPV) calculated to quantify the diagnostic performance of statuses of each potential biomarker (MSS/MSI and RNF43 separated, and MSS/MSI-RNF43 combined, in the discovery and validation cohorts). Abbreviations: MSI, microsatellite instability high; MSS, microsatellite stable.
Extended Data Fig. 5 Waterfall plots depicting different molecular subtypes.
A) Integrated waterfall plot showing best change in the total diameter of target lesions from baseline in 86 patients with mCRCBRAF-V600E tumors treated with anti-BRAF/EGFR combinatory therapies (discovery cohort, n = 44, plus validation cohort, n = 42). As a common in real-world clinical practice for aggressive tumors like mCRCBRAF-V600E, images from a proportion of patients (n = 12) were not available, mostly due to rapid clinical deterioration. B–D) Waterfall plots including patients with the different molecular subtypes: B) MSI-RNF43mutated tumors are shown in gray, C) MSS-RNF43wild-type tumors are shown in yellow, and D) MSS-RNF43mutated tumors are shown in blue. E) The post-contrast CT scan at baseline shows multiple liver metastases (yellow arrows) in a patient with mCRCBRAF-V600E with a MSS-RNF43mutated tumor. F) The post-contrast CT scan after 3 months of treatment shows marked reduction in size of the liver metastases (yellow arrows); some of the lesions are no longer seen in the follow-up CT-scan (yellow circles). Abbreviations: MSI, microsatellite instability high; MSS, microsatellite stable.
Extended Data Fig. 6 Kaplan-Meier curves of PFS and OS from patients with mCRCBRAF-V600E tumors (discovery cohort, n = 45) treated with anti-BRAF/EGFR combinatory therapies with regard to RNF43 mutations (left) and RNF43 mutations combined with MSS/MSI status (right).
A) PFS of patients with RNF43mutated (n = 19) and RNF43wildtype (n = 26) tumors and B) combined RNF43 and MSS/MSI statuses (MSS-RNF43wild-type [n = 26], MSS-RNF43mutated [n = 11], MSI-RNF43mutated [n = 8]). C) Event-free survival of patients with RNF43mutated (n = 19) and RNF43wild-type (n = 26) tumors and D) combined RNF43 and MSS/MSI statuses (MSS-RNF43wild-type [n = 26], MSS-RNF43mutated [n = 11], MSI-RNF43mutated [n = 8]). Cox models were used to obtain hazard ratios with 95% CIs, and the two-sided log-rank test was used for statistical comparisons without adjustment for multiplicity. Colors indicate molecular subtypes: RNF43wild-type tumors with or without MSS (yellow), RNF43mutated tumors with or without MSS (blue) and MSI-RNF43mutated tumors (gray). Significant values are shown in bold. Abbreviations: CI, confidence interval; HR, hazard ratio.
Extended Data Fig. 7 Clinical characteristics and mutational profile of patients with mCRCBRAF-V600E treated with no anti-BRAF/EGFR combinatorial therapies.
A) Clinical and therapeutic characteristics of patients from the control cohort, patients receiving standard chemotherapy ± antiangiogenic agents (total N = 68). These patients received in total 135 chemotherapy regimens with/without anti-angiogenic drugs during first, second, or third lines. A total of 67 treatment lines (second and third line) in MSS patients were analyzed. B–E) Comparison of RNF43 mutation frequencies and protein location between discovery/validation (B,D) and control (C,E) cohorts. RNF43 mutation frequencies of the mCRCBRAF-V600E MSS subgroup from discovery/validation (29%, 22/76) and control (22%, 15/68) cohorts were similar (B, C), respectively. Also, the MSS discovery/validation and control cohorts showed a similar RNF43 mutational profile regarding localization and predictive functional status (D, E). Abbreviations: MSS, microsatellite stable.
Extended Data Fig. 8 Analysis of the prognostic/predictive value of RNF43 mutations in the control cohort of patients with mCRCBRAF-V600E with MSS tumors treated with standard-of-care regimens only, with no exposure to anti-BRAF therapies.
A, B) Kaplan-Meier curves of PFS and OS from the control cohort treated in second or third line (n = 67) C, D) and in first line (n = 68) with standard-of-care regimens only, with no exposure to anti-BRAF therapies. Colors refer to molecular subtypes: RNF43wild-type (yellow), RNF43mutated tumors (blue)wild-type. Cox models were used to obtain hazard ratios with 95% CI, and the two-sided log-rank test was used for statistical comparisons without adjustment for multiplicity. Abbreviations: CI, confidence interval; HR, hazard ratio.
Extended Data Fig. 9 Mutation frequencies of RNF43 and APC genes in MSS status and immunohistochemical analysis against human β-catenin of mCRCBRAF-V600E tumor samples.
A, B) Mutation frequencies of RNF43 and APC genes, and response status from patients with mCRCBRAF-V600E with MSS tumors from the discovery (A) and validation (B) cohorts. Chi-square test was used for the statistical analysis. Abbreviations: CR, complete response; MSS, microsatellite stable; PD, progressive disease; PR, partial response; SD, stable disease. C–D) Immunohistochemical analysis against human β-catenin was performed in MSS-RNF43wild-type, MSS-RNF43mutated, and MSI-RNF43mutated mCRCBRAF-V600E tumor samples (n = 33) to search for potential differential pattern in total expression or cellular localization of β-catenin protein. No correlation was found between β-catenin protein expression levels (0, no staining; 1, weak; 2, moderate; and 3, strong) and cellular localization (cytoplasm, membrane, nucleus) with response to anti-BRAF/EGFR ± combinatorial therapies. Representative immunohistochemistry for β-catenin with strong membrane positivity, weak-moderate cytoplasmic positivity, and few weak nucleus positivity (magnification 40x) (C) and strong-moderate nuclear and cytoplasmic positivity (D) (magnification 40x).
Supplementary information
Supplementary Tables
Supplementary Table 1: Clinical and genetic dataset of mCRCBRAF-V600E from the discovery and validation cohorts. Supplementary Table 2: Comparison of mutation frequencies of cancer-related genes from the discovery cohort and patients with mCRCBRAF-V600E from the TCGA PanCancer Atlas.
Source data
Source Data Fig. 6
Original unprocessed western blot from Fig. 6c.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elez, E., Ros, J., Fernández, J. et al. RNF43 mutations predict response to anti-BRAF/EGFR combinatory therapies in BRAFV600E metastatic colorectal cancer. Nat Med 28, 2162–2170 (2022). https://doi.org/10.1038/s41591-022-01976-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-022-01976-z
This article is cited by
-
Evolutionary dependency of cancer mutations in gene pairs inferred by nonsynonymous-synonymous mutation ratios
Genome Medicine (2024)
-
Molecular profiling of BRAF-V600E-mutant metastatic colorectal cancer in the phase 3 BEACON CRC trial
Nature Medicine (2024)
-
BRAF — a tumour-agnostic drug target with lineage-specific dependencies
Nature Reviews Clinical Oncology (2024)
-
Mechanisms of metastatic colorectal cancer
Nature Reviews Gastroenterology & Hepatology (2024)
-
Molecular profiling of pediatric and young adult colorectal cancer reveals a distinct genomic landscapes and potential therapeutic avenues
Scientific Reports (2024)
