
Abstract
Interlinked and fundamental aging processes appear to be a root-cause contributor to many disorders and diseases. One such process is cellular senescence, which entails a state of cell cycle arrest in response to damaging stimuli. Senescent cells can arise throughout the lifespan and, if persistent, can have deleterious effects on tissue function due to the many proteins they secrete. In preclinical models, interventions targeting those senescent cells that are persistent and cause tissue damage have been shown to delay, prevent or alleviate multiple disorders. In line with this, the discovery of small-molecule senolytic drugs that selectively clear senescent cells has led to promising strategies for preventing or treating multiple diseases and age-related conditions in humans. In this Review, we outline the rationale for senescent cells as a therapeutic target for disorders across the lifespan and discuss the most promising strategies—including recent and ongoing clinical trials—for translating small-molecule senolytics and other senescence-targeting interventions into clinical use.
Similar content being viewed by others
Main
The aging population is steadily increasing. The World Health Organization (WHO) estimates that 1 in 6 people, or 2.1 billion, are expected to be over age 60 by 2030 (ref. 1). Chronological age is the major predictor for most of the diseases that account for the bulk of morbidity, mortality and health costs across low-, middle- and high-income countries2,3,4,5,6,7.
Aging progresses throughout the lifespan can be accentuated at etiologic sites of multiple acute and chronic diseases, including in children6,8,9,10,11,12. Indeed, fundamental aging processes can operate even before conception, for example, in the context of aged oocytes linked to Down syndrome12,13. A fundamental aging mechanism that has gained increasing attention is cellular senescence. Senescent cells accumulate during aging and at pathogenic sites of multiple disorders and diseases. After the first reports of senolytic drugs (agents that selectively eliminate senescent cells) in 2015 (ref. 14), promising results from preclinical studies have facilitated progression to early-phase clinical trials evaluating the safety and efficacy of senolytics, some of which have now been published (Table 1).
Fundamental aging mechanisms can be grouped into so-called hallmarks or ‘pillars’ of aging; these include genomic instability, progenitor cell exhaustion/dysfunction, telomeric and epigenetic changes, dysregulated protein homeostasis, altered nutrient sensing, mitochondrial dysfunction, altered intercellular communication, chronic low-grade inflammation, fibrosis, microbiome dysregulation and cellular senescence3,15. The Geroscience Hypothesis holds that these pillars of aging, including cellular senescence, tend to progress in concert and may be root-cause contributors to the pathophysiology of multiple diseases, age-related dysfunction (including the geriatric syndromes such as frailty, immobility, sarcopenia/muscle wasting, mild cognitive impairment and incontinence) and loss of resilience (for example, decreased ability to recover from stresses such as injury, surgery, chemotherapy or infections or to mount an antibody response to immunizations)3,15,16,17,18. The Unitary Theory of Fundamental Aging Mechanisms builds on the Geroscience Hypothesis by positing that interventions targeting any one fundamental mechanism may target the others6. For example, interventions that target cellular senescence tend to attenuate other fundamental aging mechanisms leading to reduced inflammation, attenuated exhaustion of progenitors, decreased fibrosis, alleviated mitochondrial dysfunction and a partially restored microbiome in experimental animal models of aging and chronic diseases6,7,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36.
By understanding and targeting cellular senescence and the other pillars of aging, rather than targeting individual diseases that are downstream of fundamental aging processes, it is conceivable that multimorbidity could be reduced and healthspan increased, with realization of substantial societal and economic benefits4,6. In this Review, we consider the potential value of senescent cells as a therapeutic target, the current state of senolytic drug development and the path to bring preventive and therapeutic strategies targeting senescent cells to the clinic.
Cellular senescence: mechanisms and pathways
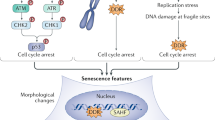
Cellular senescence was first reported in 1961 by Hayflick and Moorhead after serially subculturing human fibroblasts37. Senescent cells, which are in a state of essentially irreversible cell cycle arrest but remain viable, can accumulate with aging, especially in more frail individuals, and at pathogenic sites of multiple disorders and diseases in experimental animals and humans across the lifespan. The senescent cell fate can be triggered by a number of stressors including DNA damage, cancerous mutations or oncogene activation, mitochondrial dysfunction, reactive metabolites, hyperoxia or hypoxia, proteotoxic stress, extracellular signals, infections, mechanical or shear stresses that deform cells, resistance exercise and factors secreted by other senescent cells18,38,39,40,41,42,43,44,45,46,47. Many such stressors activate DNA damage response signaling and activation of the p53/p21CIP1/WAF1, p16INK4a/retinoblastoma protein or other pathways, resulting in cell cycle arrest and the development of a senescence-associated secretory phenotype (SASP)24,40,48,49,50,51,52,53 (Fig. 1). Through upregulation of pro-survival and antiapoptotic pathways such as SRC kinases, the PI3K–AKT signaling pathway, heat shock protein (HSP) pathways, serpines, mitochondrial pathways or apoptosis regulator BCL-2-related proteins, those senescent cells with a proapoptotic SASP can survive, despite the cytotoxic microenvironment they create6,7,14,34,35,54,55,56,57,58.

The SASP is a key feature of cellular senescence. Cellular stressors induce DNA damage response signaling, which activates key transcription factors and pathways including NF-κB, CCAAT/enhancer binding protein-β (C/EBPβ), GATA binding protein 4 (GATA4), p38 and JAK–STAT, which can drive and modulate the SASP31,48,60,63,64,103,157,214,215,216,217,218. The various forms of the SASP can comprise chemokines, extracellular matrix proteases, remodeling factors, bioactive lipids, noncoding nucleotides and reactive metabolites7,31,59,60,61,62,63,64. IL-6, interleukin-6; ROS, reactive oxygen species; TGF-β, transforming growth factor beta; TIMPs, tissue inhibitors of metalloproteinases; TNF, tumor necrosis factor.
The senescence-associated secretory phenotype
Most cells undergoing senescence develop a SASP (Fig. 1). In 30–70% of senescent cells, this SASP entails pro-inflammatory, proapoptotic and pro-fibrotic factors7,31,40,59,60,61,62,63,64, some of which can cause previously non-senescent cells to become senescent both locally and at a distance in an endocrine manner24,39,47. This proapoptotic SASP can have detrimental effects both locally and systemically due to senescent cell accumulation and persistence24. In the other 30–70% of senescent cells, the SASP appears to comprise growth and other regenerative factors, potentially causing less apoptosis, tissue destruction and fibrosis than proapoptotic, pro-inflammatory senescent cells and can even contribute to tissue repair (for example, through the growth factors VEGF-A and PDGF-AA)61,65.
If senescent cells are present transiently, beneficial functions of both the proapoptotic and pro-growth types of SASP can orchestrate tissue remodeling (during fetal development, growth of younger individuals, and after cell or tissue damage), induce immune responses during infections or tissue injury, promote parturition by SASP factors released by placental senescent cells, and induce clearance of those senescent cells with a pro-inflammatory SASP because they attract, anchor and activate immune cells10,11,14,61,62,65.
Adverse impacts of persistent, proapoptotic SASP-expressing senescent cells
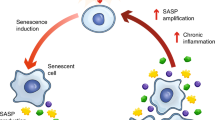
Normally, senescent cells appear to be cleared within days to weeks after they develop by natural killer cells and other immune cell types62,66,67,68,69. However, if a threshold burden of senescent cells is exceeded, senescent cells can accumulate, perhaps because proapoptotic SASP-expressing senescent cells induce paracrine and endocrine spread of senescence at a rate exceeding immune clearance of preexisting and newly formed senescent cells24,62 (Fig. 2). Once senescent cell burden surpasses this threshold, continuing increases in proapoptotic/pro-inflammatory senescent cell burden may contribute to tissue destruction and hence development or progression of multiple diseases and age-related disorders (Table 2) as well as immune dysregulation, further amplifying senescent cell accumulation in a feed-forward loop36,38,62. Although not every senescent cell develops a proapoptotic, inflammatory SASP, accumulation and persistence of such senescent cells can induce a chronic low-grade, pro-fibrotic inflammatory state (usually associated with aging and chronic diseases), known as ‘inflammaging’70. This sterile inflammatory state can provoke dysfunction of neighboring and distant non-senescent cells, such as progenitor cells, contributing to impaired tissue function and reduced regenerative capacity18,21,24,26,34. Consistent with this, persistence of senescent cells has been implicated in causing disorders related to tissue inflammation, fibrosis and extracellular matrix degradation, adipose tissue insulin resistance, reduced muscle hypertrophy after resistance exercise and impaired fracture repair in older individuals, as well as promoting malignant transformation6,18,22,23,48,71,72,73,74,75,76,77.

This theory postulates that once senescent cell burden exceeds a threshold, self-amplifying paracrine and endocrine spread of senescence through the SASP outpaces clearance of senescent cells by the immune system34,219. Additionally, increased abundance of SASP factors may impede immune system function62,78, further amplifying accumulation of senescent cells. Senescent cell accumulation may also accelerate other fundamental aging mechanisms. In studies of effects of transplanting senescent versus non-senescent cells into middle-aged mice, a minimum number of transplanted senescent cells was necessary to cause accelerated aging-like phenotypes24. In conditions in which senescent cell burden is already high, such as obesity, fewer senescent cells need to be transplanted to induce the same effect as in lean mice of the same age23,24,151. Consistent with this, in human childhood cancer survivors who have had DNA-damaging anticancer therapy, a subsequent accelerated aging-like phenotype can occur at a considerably earlier age than in older individuals who do not have a history of childhood cancer treatment169. Hence, senescent cells with a proapoptotic, inflammatory SASP may need to exceed a threshold to exert detrimental effects. Systemic clearance of senescent cells by genetic or pharmacologic means tends to attenuate the other pillars of aging and can delay, prevent or alleviate multiple age-related disorders and diseases23,24,30,49.
The SASP of senescent cells is not static; it can change over time78,79,80 and varies depending on the type of cells that became senescent and how senescence was induced16,44,59,60,81. The intracellular and extracellular environment can modulate which SASP factors are produced and their abundance. Persistent senescent cells appear to be highly responsive to extracellular cues, such as damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), which can exacerbate the proapoptotic, pro-inflammatory qualities of the SASP16. For example, in the case of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, some of these extracellular cues are mediated through Toll-like receptor 3 and angiotensin converting enzyme-2, contributing to coronavirus disease 2019 (COVID-19) morbidity44,82. Intracellular cues can exacerbate damaging, pro-inflammatory properties of the SASP several weeks or months after senescence has been induced. These include retrotransposable elements (for example, LINE-1), cytosolic mitochondrial DNA, or circular DNA—all of which can activate the cytosolic DNA-sensing (cGAS)–STING pathway, triggering the expression of pro-inflammatory genes79,80,81.
Discovery and development of senolytic drugs
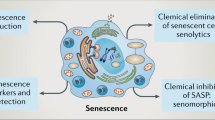
The first senolytic drugs were identified using a hypothesis-driven, mechanism-based drug discovery approach (Fig. 3). Because those 30–70% of senescent cells that have a proapoptotic, tissue-destructive SASP are themselves resistant to apoptosis, it was hypothesized that such senescent cells depend on antiapoptotic, pro-survival pathways to avoid self-destruction14,83. Analysis of proteomic and transcriptomic datasets revealed there is indeed upregulation of one or more senescent cell antiapoptotic pathways (SCAPs) in senescent cells. SCAP pathways are similar to those that protect certain types of cancer cells, such as B cell lymphoma or chronic lymphocytic leukemia cells, which also release tissue-destructive proapoptotic factors but evade undergoing apoptosis themselves84,85. Transiently disabling SCAP pathways results in apoptosis of the senescent cells with a tissue-destructive SASP, while non-senescent cells or those senescent cells with a pro-growth, non-apoptotic SASP remain viable (U. Tripathi, S.C., L.G.P.L. Prata, T.T. and J.L.K., unpublished data).

First-generation senolytics target different SCAPs, including tyrosine kinase receptors (TKRs), growth factor receptors (GFRs), ephrin receptor B1 (EFNB1), SRC kinases, PI3K–AKT, HSP90, BCL-2 family members, caspase inhibition and p53 modulation14,54,57,86,87,88. High-throughput library screens and other approaches have informed second-generation senolytic strategies, including lysosomal and SA-β-gal-activated prodrugs and nanoparticles54,92,93,220, sodium–potassium pump (Na+/K+-ATPase)-dependent apoptosis194,221, SASP inhibition103,104,105,106 and immune-mediated clearance by CAR T cells, antibody–drug conjugates or vaccines62,96,97,98,99.
Bioinformatic analyses identified 46 compounds that target SCAP pathways as being potentially senolytic14. The first senolytic agents intentionally selected for further investigation were ones that: (1) target several SCAPs, rather than adhering to the traditional drug development approach of one drug/one molecular target/one disease, (2) can be administered orally, and (3) are natural products with known safety profiles or are already approved by the US Food and Drug Administration (FDA) for other indications, to facilitate translation from bench to bedside. These included the SRC/tyrosine kinase inhibitor dasatinib (D), which has been approved and extensively used since 2006 and has a quite good safety profile, and the natural flavonoids quercetin (Q) and fisetin (F), which are present in fruits and other foods14,86.
In some types of senescent cells, SCAPs can be redundant, so that targeting a single SCAP may not eliminate such cells—but combination treatment targeting multiple SCAPS may be effective. As an example, senescent mesenchymal embryonic fibroblasts from Ercc1−/− mice and bone marrow mesenchymal progenitors from old mice are not eliminated by either D or Q alone, but are eliminated by the combination of these agents14. Consistent with the heterogeneity of SCAPs across different senescent cell types, senescent human fat cell progenitors (preadipocytes or mesenchymal stromal cells) are sensitive to D but not Q or F, while senescent human umbilical vein endothelial cells are sensitive to Q or F but not D14. Since the first SCAPs were discovered, others have been identified and, based on these, more senolytic strategies have been developed. For example, forkhead box O4 (FOXO4) retains p53 in the nucleus, so peptides interfering with this interaction can lead to p53-mediated apoptosis in some types of senescent cells87. HSP90 prevents proteasomal degradation of AKT, hence inhibiting HSP90 disables pro-survival signaling through this SCAP node and results in elimination of some senescent cell types54. Consistent with this, certain HSP90 inhibiting drugs, such as geldanamycin, are senolytic against particular senescent cell types.
In some cases, senolytic compounds that target a single SCAP node, such as the BCL-2 pathway inhibitors, N (ABT-263), A1331852 or A1155463, tend to induce apoptosis in a restricted range of senescent cell types57,86. However, it is worth noting that N can cause thrombocytopenia and neutropenia, even after brief exposures57,88,89,90,91; this raises the question of whether agents that target a single molecular pathway have a greater risk of toxicity due to off-target effects associated with the high dosing required to fully suppress a single SCAP node. Perhaps by using agents or combinations that ‘lightly’ impact a number of nodes, it may be feasible to target a broader range of senescent cell types while using lower doses of each agent, thereby potentially improving the side-effect profiles of these agents. The latter approach has been used to improve tolerability of antibiotic treatments, for example.
Second-generation senolytics are now being identified using high-throughput library screens and other approaches54. One approach stems from the increase in lysosomal mass and senescence-associated β-galactosidase activity in many senescent cells, whereby galacto-oligosaccharide-coated nanoparticles and β-galactosidase-activated prodrugs appear to eliminate at least some of these cells92,93. Another approach is based on the high lysosomal activity of some senescent cells that renders them sensitive to lysosomal ATPase inhibitors94. Due to ruptured lysosomal membranes, at least some senescent cells depend on glutamine metabolism as a pH-buffering system, inhibition of which renders them vulnerable to apoptosis95.
Other strategies for decreasing age-related senescent cell burden and pathologic conditions involve modulating immune clearance of senescent cells62. Certain cell surface proteins tend to be more highly expressed by senescent cells than most other cell types, which prompted development of engineered chimeric antigen receptor (CAR) T cells, vaccines and antibody–drug conjugates targeting these cell surface markers. Each of these approaches eliminates senescent cells, although in some cases, activated macrophages and other non-senescent cell types are also affected96,97,98,99. It is not yet clear if these approaches eliminate primarily those senescent cells with a proapoptotic, inflammatory, tissue-destructive SASP, those with a mainly growth-promoting SASP, or both forms of senescent cells. A possible advantage of small-molecule senolytics over vaccines or transplanted CAR T cells is that if a need for senescent cells occurs, for example, during wound healing, tissue remodeling or pregnancy, then treatment can be discontinued—whereas the continued elimination of senescent cells induced by vaccines or CAR T strategies may not readily be switched off. Furthermore, CAR T cell therapy is expensive, generally has to be specifically formulated for each individual being treated, and can lead to graft versus host disease, necessitating prolonged immunosuppressive therapy with all its attendant risks.
It should be noted that in commonly used preclinical models, senescence is abolished by means of p16INK4a-based or p21CIP1/WAF1-based genetic clearance and this senescence-targeting approach acts through mechanisms that are distinct from first-generation, SCAP-targeting small-molecule senolytics; as a result, there appear to be differences in the types of senescent cells targeted. For example, cell types such as activated macrophages, which may not be classically senescent, can have high p16INK4a expression100, but are not targeted by D + Q24. Unpublished work from our own laboratory suggests that there may be differences in the phenotypic effects (such as those relating to wound healing and healthspan) of targeting senescent cells in transgenic mice compared to the effects of senolytic agents in wild-type mice. This is in agreement with a recent report showing that the removal of senescent cells expressing high levels of p16INK4a can lead to fibrosis in mice101, whereas small-molecule senolytics appear to reduce fibrosis in several mouse tissues22,71,73,75,102. Indeed, work to develop senolytics began before genetic models of senescent cell clearance were published and did not depend on those models34,56. These genetic models have been useful in pinpointing those cells expressing particular senescence-linked markers (for example, p16INK4a or p21CIP1/WAF1) and as complementary tools in senolytic proof-of-concept studies (Table 2). However, given their inability to eliminate the naturally occurring heterogeneous senescent cell pool (that is, elimination of both p16INK4a-expressing or p21CIP1/WAF1-expressing cells or senescent cells that express neither) and the fact that also non-senescent cells such as activated macrophages are targeted100, these genetic models are of limited use to assess the translational potential of senolytic agents—but are useful for mechanistic insights nonetheless.
SASP inhibitors
Suppressing the SASP without eliminating senescent cells is an alternative therapeutic approach for alleviating cellular senescence-related phenotypes or diseases. SASP inhibitors (senomorphics) can directly or indirectly attenuate the SASP of senescent cells by inhibiting transcription factor nuclear factor (NF)-κB, the JAK–STAT signal transduction pathway, the serine/threonine protein kinase mTOR, mitochondrial complex-1-related or 4-related targets, or other pathways involved in the induction and maintenance of the SASP103,104,105,106. In vitro and in vivo, inhibitors of NF-κB (mediating the cell response to inflammation), can reduce pro-inflammatory SASP cytokines and chemokines104. In addition, an RNA-mediated interference screen revealed that targeting alternative splicing in senescent cells may be a viable approach for inhibiting the SASP107. Rapamycin and its analogs (so-called ‘rapalogs’) suppress the SASP by inhibiting mTOR and appear to extend healthspan and lifespan in mice105,108,109. The antidiabetic drug metformin, which, among other activities, inhibits the SASP, alleviates several age-related conditions and chronic diseases110,111,112,113,114. A clinical trial (TAME, Targeting Aging with Metformin) is planned to test if metformin delays the time for a second age-related disease to occur in patients who already have a single age-related condition115.
Advantages and disadvantages of senolytics versus SASP inhibitors
There are important differences between SASP inhibitors and senolytics. Whereas SASP inhibitors potentially suppress both the growth-promoting as well as the proapoptotic, inflammatory, tissue-destructive sets of SASP factors, the first-generation senolytics target the underlying cause of detrimental SASP factor production by eliminating those senescent cells that release proapoptotic factors. In the case of SASP inhibitors, continuous treatment is needed to maintain suppression of the SASP, although some agents, such as rapamycin, can have prolonged effects after a brief course of administration116,117. This may be a result of inhibiting SASP-mediated spread of senescence, thereby allowing the innate and adaptive immune system to further reduce senescent cell burden, consistent with the Unitary Theory (Fig. 2). With senolytics, intermittent administration appears to be as effective as continuous treatment for attenuating senescent cell burden21. This intermittent ‘hit-and-run’ strategy of senolytic administration could serve to reduce side effects of agents such as D, which generally appear after weeks to months of continuous administration118,119. In this regard, the advantages of D, Q or F over some other senolytics are their brief half-lives (4, 11 or 3–4 h, respectively, in humans) and rapid elimination120,121,122. The greater need for continuous administration of SASP inhibitors could lead to more side effects than seen with intermittently dosed senolytics and could also lead to off-target effects due to suppression of cytokine secretion—even when such cytokines are needed—by non-senescent cells such as innate or adaptive immune cells. Furthermore, some SASP inhibitors can have agent-specific off-target effects, for example, rapamycin, which can cause nephrotoxicity, metabolic impairment and susceptibility to infections, at least at higher doses in mice109.
Senolytics cause SASP-expressing senescent cells to undergo apoptosis, and such senescent cells are present at sites of dysfunction. Interestingly, a study involving transplanted mesenchymal stromal cells, which have therapeutic effects in a wide range of disease models, may hint at a possible mechanism for the beneficial effects of senolytic-induced apoptosis. The study suggested that apoptosis of mesenchymal stromal cells is required for their therapeutic effects, possibly by means of downstream immunosuppressive effects of apoptotic processes123. This raises the intriguing hypothesis that senolytic-induced apoptosis of destructive SASP-expressing senescent cells, which are concentrated at sites of pathology, might contribute to the beneficial effects of senolytics, which has not been directly tested in preclinical models in currently available reports.
Consideration of the different cell populations affected by senolytic and SASP inhibitor interventions, whether expressing a detrimental tissue-destructive or beneficial pro-growth factor secretory phenotype, is crucial to the successful development of senotherapeutic interventions61. Elimination of all senescent cells or general inhibition of the SASP might be detrimental in some instances in which senescent cells are beneficial. However, interventions that predominantly target the persisting, tissue-destructive SASP-expressing senescent cells might have superior therapeutic potential and fewer off-target effects.
Senolytics in preclinical models
First-generation senolytics, such as D + Q, have been tested in several preclinical models of aging and diseases, including type 2 diabetes, and bone, heart, kidney, liver, lung, muscle and neurological disorders (Table 2). Whereas transplanting senescent cells decreases healthspan and lifespan in preclinical models, eliminating transplanted or endogenous senescent cells increases healthspan, thereby fulfilling Koch’s postulates for causality24,124.
In mouse models of diet-induced obesity, reducing the burden of senescent cells in adipose tissue by administering D + Q attenuates adipose tissue inflammation, alleviates metabolic dysfunction, and restores the capacity of preadipocytes to differentiate into functional, mature, insulin-responsive fat cells25. High-fat diets (HFDs) and obesity result in senescent cell accumulation and lead to impaired function of multiple organs. For example, HFD-induced senescent mouse kidney cells are linked to renal fibrosis and functional impairment; Q treatment reduces senescent cell burden and SASP marker expression, alleviates renal fibrosis and improves kidney function102. In livers of HFD-fed mice, oral D + Q reduces the burden of senescent hepatocytes and hepatic steatosis75. Obesity also leads to accumulation of senescent cells near the third ventricles of the murine brain, together with development of neuropsychiatric dysfunction, particularly anxiety, which results from altered function of the limbic system situated near the third ventricles. Oral D + Q reduces this neuroinflammation, increases markers of neurogenesis, reduces gliosis and alleviates anxiety in obese mice33. Fibrosis can be a senescence-driven progressive process that contributes to reduced organ function; in a mouse model of idiopathic pulmonary fibrosis (IPF), D + Q treatment improved lung compliance and reduced frailty71. In age-related osteoporosis linked to senescent-like osteocytes and bone marrow cells in mice, D + Q reduced development of bone-resorbing osteoclasts, while increasing differentiated bone-forming osteoblasts, with restoration of bone mass21. After resistance exercise, senescent cells develop in muscle of old mice in tandem with impaired exercise-induced muscle hypertrophy compared to young mice18. D + Q alleviates this negative impact of aging on muscle growth. Recently, senescent cells have been implicated in accentuating the severity of viral infections due to amplification of SASP factor secretion, predisposing to cytokine storm and multi-organ failure. In mice infected with a murine coronavirus, the mortality rate was reduced following administration of D + Q or F16.
Transplanted senescent cells or organs from old mice have been shown to spread the senescent phenotype to distant sites in recipient mice, predisposing to detrimental outcomes after transplantation24,31. D + Q treatment of either old donor mice, the organs harvested from the old donors before transplantation, or the recipients, led to decreased senescent cell burden and increased survival of recipient mice – so that outcomes resembled those observed in mice receiving a transplant from a young donor31. Hence, a potential new application of senolytics may entail ex vivo perfusion of organs from older donors that are currently being discarded because of increased risk of organ failure or allograft rejection, potentially offsetting shortages of organs for transplantation125.
A recent study implicated cellular senescence and the beneficial effects of senolytics for Down syndrome (trisomy 21)13. A third copy of chromosome 21 in neural progenitor cells leads to transcriptional and nuclear organizational changes similar to those in senescent cells. Interestingly, D + Q alleviated transcriptional changes and cellular dysfunction in an in vitro human neural cell Down syndrome model. An accelerated aging-like phenotype has been observed in astronauts who are exposed to radiation, high G-forces, zero gravity and cabin air quality126. Thus, senolytics might support long-distance space exploration by eliminating the senescent cells caused by cosmic and solar radiation, especially outside the van Allen radiation belts. In April 2022, effects of space travel on senescence biomarkers in astronauts and cultured human fat cell progenitors were tested during the Axiom-1 mission to the International Space Station; data are pending127.
Systemic versus local administration
According to the threshold theory of cellular senescence, senescent cells can be present without clinical manifestations but, if their abundance exceeds a threshold, senescent cell burden can increase further and contribute to development of local and systemic dysfunction and multiple diseases (Fig. 2). Senescent cells can spread even to distant sites because of their SASP; therefore, systemic, intermittent administration of senolytics for senescence-associated diseases is potentially more promising than local administration, with the exception of perhaps eye, skin or dental topical or other local applications34. Perhaps there may not need to be strict cell type specificity of senolytics administered in vivo. Arguably, once systemic senolytic treatment has reduced overall senescent cell burden to below a threshold, the immune system might clear remaining senescent cells, including those resistant to that particular senolytic. This could be especially important for older individuals or those with chronic diseases, who already have a high systemic senescent cell burden24,128. Strategies for systemic senolytic administration include oral or intravenous routes, with orally active senolytics generally being more accepted by patients and less expensive to administer. The timing, the particular senolytic agent used, and the age, sex, and other characteristics of the individual may impact effectiveness of senolytics24. For example, F may be beneficial but D detrimental in healthy young female mice that have not yet accumulated many senescent cells129.
Clinical trials and future directions
Based on promising results in preclinical models, over 20 clinical trials of senolytic therapies are completed, ongoing or planned34 (Table 1). Because side effects of senolytics in humans are not yet fully known, and to maximize benefit–risk ratios, the first clinical trials are underway in patients with serious health conditions, such as diabetic kidney disease, Alzheimer’s disease, frailty and IPF34. The first in-human trial of senolytics (D + Q), the Hematopoietic Stem Cell Transplant Survivors Study, is still underway (NCT02652052; first patient dosed on 1 April 2016). The first senolytic clinical trial published was an open-label pilot study in which 14 patients with IPF were treated with intermittent D + Q on 3 d per week for 3 weeks63. Results suggested that senolytics improved physical function in these frail patients. Furthermore, post hoc analysis of a study involving 20 patients with IPF showed that urine levels of the ‘geroprotective’ factor α-Klotho were higher after oral D + Q than before treatment19. In an open-label phase 1 pilot study in 9 patients with diabetic kidney disease, a 3-d course of oral D + Q was sufficient to decrease adipose tissue senescent cell burden, inflammation, fibrosis and circulating SASP factors for at least 11 d after the last dose of senolytics, indicating target engagement and suggesting that an intermittent dosing regimen may be effective in humans48. These early data warrant evaluation in larger randomized, double-blind, placebo-controlled trials for senescence-associated disorders and diseases, some of which are underway34 (Table 1).
It should be noted that a phase 2, randomized, double-blind, placebo-controlled clinical trial (NCT04349956)—in which the senolytic agent was a p53-destabilizing protein MDM2 inhibitor, UBX0101 (also known as nutlin-3a)—did not achieve its primary endpoint of improving pain in patients with osteoarthritis of the knee in a 12-week follow-up. In our hands and others, nutlin-3a does not show or shows only weak senolytic activity and, in some cases, can even induce cellular senescence95,130. Furthermore, in a mouse model of osteoarthritis, a combination of UBX0101 with N was necessary to restore aged joint structure131. Thus, the failure of this clinical trial seems related to the particular agent administered, which may not have been a fully effective senolytic.
Biomarkers of senescent cell burden and senolysis: gerodiagnostics
Identification and monitoring of senescent cell burden in situ, especially during clinical trials to assess safety and efficacy, can be challenging if solely based on analysis of tissue biopsies given the contextual and complex regulation of the senescent cell fate. However, SASP factors, senescence markers and markers of other fundamental aging processes (for example, α-Klotho) can be assayed in body fluids such as urine, saliva, blood or cerebrospinal fluid7,19,48,59,61,64,132,133. Additionally, ongoing efforts are underway to identify and define biomarkers of senescent cell accumulation (senescence biomarkers) and destruction of senescent cells during senolytic treatment (senolysis biomarkers) that meet requirements of regulatory authorities. Panels of SASP factors and senolysis markers for clinical trials and, ultimately, clinical practice will be important as diagnostics/predictors for multiple disorders and diseases, monitoring target engagement and individualizing senolytic regimens for patients. Indeed, to facilitate developing interventions targeting fundamental aging processes and for eventual use in clinical practice, such indicators will need to be more than mere ‘biological clocks’. Indicators are needed that are reproducible, reliable, feasible and inexpensive to measure, and reflect not only biological age, but also correlate with clinical function and/or disabilities. These should also change in response to interventions, predict or reflect changes in clinical function caused by these interventions, and indicate which senolytic or SASP inhibitor may be best for an individual. Such biomarkers or composite scores of biomarkers are known as ‘gerodiagnostics’. Indeed, the first gerodiagnostic score sensitive to senolytic administration in humans was a blood composite score published in 2019 (ref. 48). Additionally, we propose the concept of ‘gerodiagnostic ratios’, whereby gerodiagnostic indicators that increase with aging or disorders linked to acceleration of fundamental aging mechanisms, could be summed into a composite in the numerator (for example, markers of senescent cells, SASP factors, CD38 and mTOR activity indicators), while beneficial geroprotective factors (for example, α-Klotho or perhaps nicotinamide adenine dinucleotide (NAD+) or Sirt-6) could be in the denominator. This approach could both enhance sensitivity and reduce ‘denominator effects’; for example, creatinine is often used as a dominator for urine analytes, introducing the confounding variable of muscle mass, upon which creatinine levels depend.
Clinical trajectory for the next decade
Large, randomized controlled trials to assess and ensure safety, benefit and target engagement of senolytics are needed to validate preliminary results from early-phase clinical trials (Table 1). If safety and effectiveness of senolytics are first demonstrated in patients with serious diseases for which current treatments are inadequate, it could become acceptable to test them for less severe senescence-linked disorders. If safe and effective for such conditions, senolytics might then be tested for prevention of age-related dysfunction and diseases in older individuals, using a strategy like that which is planned for metformin in the TAME study115. TAME will test if metformin delays the appearance of a second age-related disorder in patients who already have one such disorder; it will not be a trial including completely healthy older adults115. If attempts to extend human healthspan are effective, future studies might conceivably aim to evaluate the role of senolytic therapies in extending human lifespan.
Combination therapeutic strategies
Clinical studies of geroscience interventions targeting the other pillars of aging are currently underway or being planned. Dietary interventions, such as caloric restriction and intermittent fasting, might be protective against development of age-related dysfunction and diseases based on studies in animal models134,135. Exercise may delay senescent cell formation and reduce inflammation, frailty and chronic disease onset in mice136,137. Metformin, resveratrol and rapalogs (agents related to rapamycin) are SASP inhibitors and appear to impact some of the same basic processes as caloric restriction or exercise110,138. Sirtuins facilitate DNA damage repair, partially relying on oxidized NAD+ to do so. Senescent cells can decrease NAD+ through SASP activation of the NAD-degrading enzyme CD38 on the surface of macrophages and, remarkably, senolytics partially restore tissue NAD+ levels139,140. NAD+ or NAD precursors can increase healthspan in experimental animals141. The non-feminizing estrogen, 17α-estradiol, declines with aging in females and males and 17α-estradiol treatment extends healthspan and alleviates age-related metabolic dysfunction and inflammation in mice142. As considered above, senolytics can increase α‐Klotho, which is geroprotective, neuroprotective and linked to healthspan in mice19. Interestingly, α‐Klotho overexpression in mice increases healthspan and lifespan by up to 30% (ref. 143). Hence, consistent with the Unitary Theory, interventions targeting the different fundamental aging mechanisms appear to alleviate aging phenotypes, delay, prevent or treat multiple diseases, and extend healthspan in preclinical models. Testing combinations of these lifestyle, nutritional, natural product and pharmacologic interventions may be informative. However, if the Unitary hypothesis is correct, they may contribute less-than-additive effects because fundamental aging mechanisms are tightly interconnected. A more promising strategy could be to combine geroscience with disease-specific interventions.
Conclusions
The elimination of senescent cells has emerged as a plausible therapeutic strategy for preventing, delaying or alleviating multiple diseases and age-related dysfunction. Promising results of senolytics in preclinical models suggest therapeutic and preventive opportunities for delaying multimorbidity and increasing healthspan. While randomized controlled trials will define the safety and potential benefits of senolytic strategies, scientific and regulatory challenges must be addressed in the near term if senolytics are to be used in the clinic (Box 1).
A key priority should be the identification of reliable, sensitive and specific gerodiagnostics—biomarkers to quantify senescent cell abundance, the SASP and senolysis as well as other pillars of aging. Interventions modulating the SASP (including those that specifically upregulate or downregulate tissue-destructive factors versus growth factors) or topical senolytic agents, especially those that eliminate senescent cells with a proapoptotic SASP, could accelerate wound healing, a possibility that needs to be experimentally tested and correlated with predictive gerodiagnostics.
The lack of WHO International Classification of Disease (ICD) codes for multimorbidity, sarcopenia, healthspan or the geriatric syndromes represents a barrier to clinical development. Such ICD codes would facilitate regulatory approvals, recording of conditions linked to fundamental aging processes in hospital and insurance records, epidemiological studies, physician and hospital reimbursement and engagement of the pharmaceutical industry. The possible interdependencies among fundamental aging mechanisms need to be investigated to deepen our knowledge about basic aging mechanisms and disease etiologies and to develop treatment strategies to reduce multimorbidity and enhance healthspan.
Finally, a note of caution is important. Even though preclinical data are promising, unless and until carefully monitored, rigorous clinical trials demonstrate safety and effectiveness of senolytics or SASP inhibitors, they should not be endorsed for the prevention or treatment of diseases over-the-counter or in clinical practice. In the meantime, the results of ongoing and planned clinical trials will yield informative data and insights into the role of cellular senescence as a therapeutic target for age-related disorders, potentially enabling translation of small-molecule senolytics into the clinic in the near future.
References
World Health Organization. Ageing and health. https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (2021).
Niccoli, T. & Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 22, R741–R752 (2012).
Kennedy, B. K. et al. Geroscience: linking aging to chronic disease. Cell 159, 709–713 (2014).
Scott, A. J., Ellison, M. & Sinclair, D. A. The economic value of targeting aging. Nat. Aging 1, 616–623 (2021).
St Sauver, J. L. et al. Risk of developing multimorbidity across all ages in an historical cohort study: differences by sex and ethnicity. BMJ Open 5, e006413 (2015).
Tchkonia, T., Palmer, A. K. & Kirkland, J. L. New horizons: novel approaches to enhance healthspan through targeting cellular senescence and related aging mechanisms. J. Clin. Endocrinol. Metab. 106, e1481–e1487 (2021).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Wissler Gerdes, E. O. et al. Cellular senescence in aging and age-related diseases: implications for neurodegenerative diseases. Int. Rev. Neurobiol. 155, 203–234 (2020).
Ness, K. K. et al. Frailty in childhood cancer survivors. Cancer 121, 1540–1547 (2015).
Munoz-Espin, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
Suvakov, S. et al. Epigenetic and senescence markers indicate an accelerated ageing-like state in women with preeclamptic pregnancies. EBioMedicine 70, 103536 (2021).
Igarashi, H., Takahashi, T. & Nagase, S. Oocyte aging underlies female reproductive aging: biological mechanisms and therapeutic strategies. Reprod. Med. Biol. 14, 159–169 (2015).
Meharena, H. S. et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 29, 116–130 (2022).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Camell, C. D. et al. Senolytics reduce coronavirus-related mortality in old mice. Science 373, eabe4832 (2021).
Hadley, E. C., Kuchel, G. A., Newman, A. B. & Workshop Speakers and Participants. Report: NIA workshop on measures of physiologic resiliencies in human aging. J. Gerontol. A Biol. Sci. Med. Sci. 72, 980–990 (2017).
Dungan, C. M. et al. Senolytic treatment rescues blunted muscle hypertrophy in old mice. Geroscience https://doi.org/10.1007/s11357-022-00542-2 (2022).
Zhu, Y. et al. Orally active, clinically translatable senolytics restore α-Klotho in mice and humans. EBioMedicine 77, 103912 (2022).
Chini, C. et al. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem. Biophys. Res. Commun. 513, 486–493 (2019).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Moncsek, A. et al. Targeting senescent cholangiocytes and activated fibroblasts with B cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 67, 247–259 (2018).
Xu, M. et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4, e12997 (2015).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Palmer, A. K. et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950 (2019).
Lewis-McDougall, F. C. et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 18, e12931 (2019).
Musi, N. et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840 (2018).
Vizioli, M. G. et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34, 428–445 (2020).
Yousefzadeh, M. et al. DNA damage—how and why we age? Elife 10, e62852 (2021).
Ogrodnik, M. et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20, e13296 (2021).
Iske, J. et al. Senolytics prevent mtDNA-induced inflammation and promote the survival of aged organs following transplantation. Nat. Commun. 11, 4289 (2020).
Saccon, T. D. et al. Senolytic combination of dasatinib and quercetin alleviates intestinal senescence and inflammation and modulates the gut microbiome in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 76, 1895–1905 (2021).
Ogrodnik, M. et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 29, 1061–1077 (2019).
Kirkland, J. L. & Tchkonia, T. Senolytic drugs: from discovery to translation. J. Intern. Med. 288, 518–536 (2020).
Tchkonia, T. & Kirkland, J. L. Aging, cell senescence and chronic disease: emerging therapeutic strategies. J. Am. Med. Assoc. 320, 1319–1320 (2018).
Lagnado, A. et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 40, e106048 (2021).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell. Res. 25, 585–621 (1961).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Chaib, S., Tchkonia, T. & Kirkland, J. L. in Handbook of Experimental Pharmacology (eds Eckel, J. & Clément, K.) 165–180 (Springer, 2021).
Wiley, C. D. & Campisi, J. From ancient pathways to aging cells—connecting metabolism and cellular senescence. Cell Metab. 23, 1013–1021 (2016).
Tripathi, U. et al. SARS-CoV-2 causes senescence in human cells and exacerbates the senescence-associated secretory phenotype through TLR-3. Aging 13, 21838–21854 (2021).
Lee, S. et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289 (2021).
Martini, H. & Passos, J. F. Cellular senescence: all roads lead to mitochondria. FEBS J. https://doi.org/10.1111/febs.16361 (2022).
Tsuji, S. et al. SARS-CoV-2 infection triggers paracrine senescence and leads to a sustained senescence-associated inflammatory response. Nat. Aging 2, 115–124 (2022).
Hickson, L. J. et al. Corrigendum to ‘Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease’ EBioMedicine 47 (2019) 446–456. EBioMedicine 52, 102595 (2020).
Wang, B. et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 1, 962–973 (2021).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Yousefzadeh, M. J. et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19, e13094 (2020).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015).
Fuhrmann-Stroissnigg, H. et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422 (2017).
Xu, Q. et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat. Metab. 3, 1706–1726 (2021).
Wissler Gerdes, E. O., Zhu, Y., Tchkonia, T. & Kirkland, J. L. Discovery, development, and future application of senolytics: theories and predictions. FEBS J. 287, 2418–2427 (2020).
Zhu, Y. et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of antiapoptotic factors. Aging Cell 15, 428–435 (2016).
Gasek, N. S., Kuchel, G. A., Kirkland, J. L. & Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 1, 870–879 (2021).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118 (2010).
Tripathi, U., Misra, A., Tchkonia, T. & Kirkland, J. L. Impact of senescent cell subtypes on tissue dysfunction and repair: importance and research questions. Mech. Ageing Dev. 198, 111548 (2021).
Prata, L., Ovsyannikova, I. G., Tchkonia, T. & Kirkland, J. L. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin. Immunol. 40, 101275 (2018).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019).
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J. & Kirkland, J. L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 (2013).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Pereira, B. I. et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 10, 2387 (2019).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Brighton, P. J. et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. Elife 6, e31274 (2017).
Stokes, K. L. et al. Natural killer cells limit the clearance of senescent lung adenocarcinoma cells. Oncogenesis 8, 24 (2019).
Franceschi, C. & Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69, S4–S9 (2014).
Schafer, M. J. et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 8, 14532 (2017).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077 (2001).
Cavalcante, M. B. et al. Dasatinib plus quercetin prevents uterine age-related dysfunction and fibrosis in mice. Aging 12, 2711–2722 (2020).
Kim, S. R. et al. Transplanted senescent renal scattered tubular-like cells induce injury in the mouse kidney. Am. J. Physiol. Renal Physiol. 318, F1167–F1176 (2020).
Ogrodnik, M. et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8, 15691 (2017).
Wissler Gerdes, E. O., Misra, A., Netto, J. M. E., Tchkonia, T. & Kirkland, J. L. Strategies for late phase preclinical and early clinical trials of senolytics. Mech. Ageing Dev. 200, 111591 (2021).
Saul, D. et al. Modulation of fracture healing by the transient accumulation of senescent cells. Elife 10, e69958 (2021).
Hoare, M. et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 18, 979–992 (2016).
De Cecco, M. et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019).
Zhao, Y. et al. Transposon-triggered innate immune response confers cancer resistance to the blind mole rat. Nat. Immunol. 22, 1219–1230 (2021).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021).
Kandhaya-Pillai, R. et al. TNF-α/IFN-γ synergy amplifies senescence-associated inflammation and SARS-CoV-2 receptor expression via hyper-activated JAK/STAT1. Aging Cell 21, e13646 (2022).
Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 55, 2284–2292 (1995).
Chen, L. S., Balakrishnan, K. & Gandhi, V. Inflammation and survival pathways: Chronic lymphocytic leukemia as a model system. Biochem. Pharmacol. 80, 1936–1945 (2010).
Bessler, H. et al. Factor(s) released from irradiated B-CLL cells induce apoptosis in leukemic lymphocytes. Cancer Lett. 179, 103–108 (2002).
Zhu, Y. et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 9, 955–963 (2017).
Baar, M. P. et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147 (2017).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Wilson, W. H. et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11, 1149–1159 (2010).
Afreen, S. et al. BCL-XL expression is essential for human erythropoiesis and engraftment of hematopoietic stem cells. Cell Death Dis. 11, 8 (2020).
Josefsson, E. C., Vainchenker, W. & James, C. Regulation of platelet production and lifespan: role of Bcl-xL and potential implications for human platelet diseases. Int. J. Mol. Sci. 21, 7591 (2020).
Munoz-Espin, D. et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 10, e9355 (2018).
Guerrero, A. et al. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 19, e13133 (2020).
Dorr, J. R. et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425 (2013).
Johmura, Y. et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270 (2021).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Yoshida, S. et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 11, 2482 (2020).
Suda, M. et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat. Aging 1, 1117–1126 (2021).
Poblocka, M. et al. Targeted clearance of senescent cells using an antibody-drug conjugate against a specific membrane marker. Sci. Rep. 11, 20358 (2021).
Hall, B. M. et al. p16Ink4a and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 9, 1867–1884 (2017).
Grosse, L. et al. Defined p16high senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99 (2020).
Kim, S. R. et al. Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl. Res. 213, 112–123 (2019).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Tilstra, J. S. et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 2601–2612 (2012).
Lamming, D. W., Ye, L., Sabatini, D. M. & Baur, J. A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Invest. 123, 980–989 (2013).
Moiseeva, O. et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 12, 489–498 (2013).
Georgilis, A. et al. PTBP1-mediated alternative splicing regulates the inflammatory secretome and the pro-tumorigenic effects of senescent cells. Cancer Cell 34, 85–102 (2018).
Harrison, D. E. et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009).
Neff, F. et al. Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Invest. 123, 3272–3291 (2013).
Novelle, M. G., Ali, A., Dieguez, C., Bernier, M. & de Cabo, R. Metformin: a hopeful promise in aging research. Cold Spring Harb. Perspect. Med. 6, a025932 (2016).
Hansel, C. et al. Metformin protects against radiation-induced acute effects by limiting senescence of bronchial-epithelial cells. Int. J. Mol. Sci. 22, 7064 (2021).
Deschenes-Simard, X. et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 18, e12889 (2019).
Kulkarni, A. S., Gubbi, S. & Barzilai, N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 32, 15–30 (2020).
Fang, J. et al. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 17, e12765 (2018).
American Federation for Aging Research. Targeting the biology of aging. Ushering a new era of interventions. https://www.afar.org/tame-trial (2022).
Mannick, J. B. et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 10, eaaq1564 (2018).
Bitto, A. et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 5, e16351 (2016).
Naqvi, K. et al. Long-term follow-up of lower dose dasatinib (50 mg daily) as frontline therapy in newly diagnosed chronic-phase chronic myeloid leukemia. Cancer 126, 67–75 (2020).
Ottmann, O. et al. Long-term efficacy and safety of dasatinib in patients with chronic myeloid leukemia in accelerated phase who are resistant to or intolerant of imatinib. Blood Cancer J. 8, 88 (2018).
Christopher, L. J. et al. Metabolism and disposition of dasatinib after oral administration to humans. Drug Metab. Dispos. 36, 1357–1364 (2008).
Graefe, E. U. et al. Pharmacokinetics and bioavailability of quercetin glycosides in humans. J. Clin. Pharm. 41, 492–499 (2001).
Touil, Y. S. et al. Fisetin disposition and metabolism in mice: Identification of geraldol as an active metabolite. Biochem. Pharmacol. 82, 1731–1739 (2011).
Pang, S. H. M. et al. Mesenchymal stromal cell apoptosis is required for their therapeutic function. Nat. Commun. 12, 6495 (2021).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Tullius, S. G. & Rabb, H. Improving the supply and quality of deceased-donor organs for transplantation. N. Engl. J. Med. 378, 1920–1929 (2018).
Garrett-Bakelman, F. E. et al. The NASA twins study: a multidimensional analysis of a year-long human spaceflight. Science 364, eaau8650 (2019).
Stiepan, D. Taking Mayo Clinic research beyond the lab and into space. Mayo Clinic https://newsnetwork.mayoclinic.org/discussion/taking-mayo-clinic-research-beyond-the-lab-and-into-space (2022).
Khosla, S., Farr, J. N., Tchkonia, T. & Kirkland, J. L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 16, 263–275 (2020).
Fang, Y. et al. Sexual dimorphic responses of C57BL/6 mice to fisetin or dasatinib and quercetin cocktail oral treatment. Preprint at bioRxiv https://doi.org/10.1101/2021.11.08.467509 (2021).
Efeyan, A. et al. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 67, 7350–7357 (2007).
Faust, H. J. et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Invest. 130, 5493–5507 (2020).
Hernandez-Segura, A. et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660 (2017).
Faget, D. V., Ren, Q. & Stewart, S. A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Cancer 19, 439–453 (2019).
Madeo, F., Carmona-Gutierrez, D., Hofer, S. J. & Kroemer, G. Caloric restriction mimetics against age-associated disease: targets, mechanisms and therapeutic potential. Cell Metab. 29, 592–610 (2019).
Green, C. L., Lamming, D. W. & Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 23, 56–73 (2022).
Duggal, N. A., Niemiro, G., Harridge, S. D. R., Simpson, R. J. & Lord, J. M. Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat. Rev. Immunol. 19, 563–572 (2019).
Schafer, M. J. et al. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes 65, 1606–1615 (2016).
Baur, J. A. et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 (2006).
Chini, C. C. S. et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat. Metab. 2, 1284–1304 (2020).
Schultz, M. B. & Sinclair, D. A. Why NAD+ declines during aging: it’s destroyed. Cell Metab. 23, 965–966 (2016).
Rajman, L., Chwalek, K. & Sinclair, D. A. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 27, 529–547 (2018).
Stout, M. B. et al. 17α-estradiol alleviates age-related metabolic and inflammatory dysfunction in male mice without inducing feminization. J. Gerontol. A Biol. Sci. Med. Sci. 72, 3–15 (2017).
Kurosu, H. et al. Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833 (2005).
Spinelli, R. et al. ZMAT3 hypomethylation contributes to early senescence of preadipocytes from healthy first-degree relatives of type 2 diabetics. Aging Cell 21, e13557 (2022).
Wang, L. et al. Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 34, 75–89 (2022).
Espinosa De Ycaza, A. E. et al. Senescent cells in human adipose tissue: a cross-sectional study. Obesity 29, 1320–1327 (2021).
Salaami, O. et al. Antidiabetic effects of the senolytic agent dasatinib. Mayo Clin. Proc. 96, 3021–3029 (2021).
Conley, S. M. et al. Human obesity induces dysfunction and early senescence in adipose tissue-derived mesenchymal stromal/stem cells. Front. Cell Dev. Biol. 8, 197 (2020).
Palmer, A. K., Gustafson, B., Kirkland, J. L. & Smith, U. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia 62, 1835–1841 (2019).
Aguayo-Mazzucato, C. et al. Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 30, 129–142 (2019).
Minamino, T. et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15, 1082–1087 (2009).
Caron, M. et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 14, 1759–1767 (2007).
Yu, S. et al. Quercetin reverses cardiac systolic dysfunction in mice fed with a high-fat diet: role of angiogenesis. Oxid. Med. Cell. Longev. 2021, 8875729 (2021).
Cianflone, E. et al. Targeting cardiac stem cell senescence to treat cardiac aging and disease. Cells 9, 1558 (2020).
Dookun, E. et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 19, e13249 (2020).
Walaszczyk, A. et al. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 18, e12945 (2019).
Anderson, R. et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38, e100492 (2019).
Roos, C. M. et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016).
Childs, B. G. et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016).
Nath, K. A. et al. The murine dialysis fistula model exhibits a senescence phenotype: pathobiological mechanisms and therapeutic potential. Am. J. Physiol. Renal Physiol. 315, F1493–F1499 (2018).
Khosla, S., Farr, J. N. & Monroe, D. G. Cellular senescence and the skeleton: pathophysiology and therapeutic implications. J. Clin. Invest. 132, e154888 (2022).
Wang, H., Zhang, Q., Kaplan, F. S. & Pignolo, R. J. Clearance of senescent cells from injured muscle abrogates heterotopic ossification in mouse models of fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 37, 95–107 (2022).
Sugihara, H. et al. Cellular senescence-mediated exacerbation of Duchenne muscular dystrophy. Sci. Rep. 10, 16385 (2020).
Larsson, L. et al. Sarcopenia: aging-related loss of muscle mass and function. Physiol. Rev. 99, 427–511 (2019).
Chandra, A. et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 21, e13602 (2022).
Chandra, A. et al. Targeted reduction of senescent cell burden alleviates focal radiotherapy-related bone loss. J. Bone Miner. Res. 35, 1119–1131 (2020).
Pan, J. et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int. J. Radiat. Oncol. Biol. Phys. 99, 353–361 (2017).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017).
Cupit-Link, M. C. et al. Biology of premature ageing in survivors of cancer. ESMO Open 2, e000250 (2017).
Rahman, M. et al. Selective vulnerability of senescent glioblastoma cells to BCL-XL inhibition. Mol. Cancer Res. 20, 938–948 (2022).
Prasanna, P. G. et al. Therapy-induced senescence: opportunities to improve anticancer therapy. J. Natl. Cancer Inst. 113, 1285–1298 (2021).
Guida, J. L. et al. Strategies to prevent or remediate cancer and treatment-related aging. J. Natl. Cancer Inst. 113, 112–122 (2021).
Wang, B. et al. Transplanting cells from old but not young donors causes physical dysfunction in older recipients. Aging Cell 19, e13106 (2020).
Lau, A., Kennedy, B. K., Kirkland, J. L. & Tullius, S. G. Mixing old and young: enhancing rejuvenation and accelerating aging. J. Clin. Invest. 129, 4–11 (2019).
van Willigenburg, H., de Keizer, P. L. J. & de Bruin, R. W. F. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol. Res. 130, 322–330 (2018).
Xu, M. et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J. Gerontol. A Biol. Sci. Med. Sci. 72, 780–785 (2017).
Fatt, M. P. et al. Restoration of hippocampal neural precursor function by ablation of senescent cells in the aging stem cell niche. Stem Cell Rep. 17, 259–275 (2022).
Krzystyniak, A. et al. Combination of dasatinib and quercetin improves cognitive abilities in aged male Wistar rats, alleviates inflammation and changes hippocampal synaptic plasticity and histone H3 methylation profile. Aging 14, 572–595 (2022).
Vazquez-Villasenor, I. et al. Expression of p16 and p21 in the frontal association cortex of ALS/MND brains suggests neuronal cell cycle dysregulation and astrocyte senescence in early stages of the disease. Neuropathol. Appl. Neurobiol. 46, 171–185 (2020).
Zhang, P. et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728 (2019).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Chinta, S. J. et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940 (2018).
Govoni, S., Amadio, M., Battaini, F. & Pascale, A. Senescence of the brain: focus on cognitive kinases. Curr. Pharm. Des. 16, 660–671 (2010).
Palmer, A. K., Tchkonia, T. & Kirkland, J. L. Senolytics: potential for alleviating diabetes and its complications. Endocrinology 162, bqab058 (2021).
Kim, S. R. et al. Progressive cellular senescence mediates renal dysfunction in ischemic nephropathy. J. Am. Soc. Nephrol. 32, 1987–2004 (2021).
Mylonas, K. J. et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl. Med. 13, eabb0203 (2021).
Sfeir, J. G. & Pignolo, R. J. Pharmacologic interventions for fracture risk reduction in the oldest old: what is the evidence? JBMR Plus 5, e10538 (2021).
Novais, E. J. et al. Long-term treatment with senolytic drugs dasatinib and quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 12, 5213 (2021).
Farr, J. N. et al. Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: evidence in young adult mice and older humans. J. Bone Miner. Res. 34, 1407–1418 (2019).
Patil, P. et al. Systemic clearance of p16INK4a-positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 18, e12927 (2019).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929 (2016).
van der Feen, D. E. et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 12, eaaw4974 (2020).
Triana-Martinez, F. et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun. 10, 4731 (2019).
Barnes, P. J., Baker, J. & Donnelly, L. E. Cellular senescence as a mechanism and target in chronic lung diseases. Am. J. Respir. Crit. Care Med. 200, 556–564 (2019).
Parikh, P. et al. Hyperoxia-induced cellular senescence in fetal airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 61, 51–60 (2019).
Justice, J. N. et al. Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. J. Gerontol. A Biol. Sci. Med. Sci. 73, 939–945 (2018).
Houssaini, A. et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 3, e93203 (2018).
Nyunoya, T. et al. Cigarette smoke induces cellular senescence. Am. J. Respir. Cell Mol. Biol. 35, 681–688 (2006).
Tabibian, J. H., O’Hara, S. P., Splinter, P. L., Trussoni, C. E. & LaRusso, N. F. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 59, 2263–2275 (2014).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Florian-Rodriguez, M. E. et al. Induction of cellular senescence in rat vaginal fibroblasts and treatment with senolytics: an in vitro model for the study of pelvic organ prolapse. Female Pelvic Med. Reconstr. Surg. 28, 341–345 (2022).
Suvakov, S. et al. Targeting senescence improves angiogenic potential of adipose-derived mesenchymal stem cells in patients with preeclampsia. Biol. Sex. Differ. 10, 49 (2019).
Crespo-Garcia, S. et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 33, 818–832 (2021).
Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 77, 789–805 (2020).
Rocha, L. R. et al. Early removal of senescent cells protects retinal ganglion cells loss in experimental ocular hypertension. Aging Cell 19, e13089 (2020).
Sun, S. et al. HMGB1 and Caveolin-1 related to RPE cell senescence in age-related macular degeneration. Aging 11, 4323–4337 (2019).
Phatak, N. R., Stankowska, D. L. & Krishnamoorthy, R. R. Bcl-2, Bcl-xL and p-AKT are involved in neuroprotective effects of transcription factor Brn3b in an ocular hypertension rat model of glaucoma. Mol. Vis. 22, 1048–1061 (2016).
Fu, Q. et al. Effects of senescent lens epithelial cells on the severity of age-related cortical cataract in humans: a case–control study. Medicine 95, e3869 (2016).
Caprioli, J. Glaucoma: a disease of early cellular senescence. Invest. Ophthalmol. Vis. Sci. 54, ORSF60-7 (2013).
Liton, P. B. et al. Cellular senescence in the glaucomatous outflow pathway. Exp. Gerontol. 40, 745–748 (2005).
Nawa, N. et al. Elimination of protein aggregates prevents premature senescence in human trisomy 21 fibroblasts. PLoS ONE 14, e0219592 (2019).
Covre, L. P., De Maeyer, R. P. H., Gomes, D. C. O. & Akbar, A. N. The role of senescent T cells in immunopathology. Aging Cell 19, e13272 (2020).
Chien, Y. et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136 (2011).
Dou, Z. et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017).
Kang, C. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612 (2015).
Zhang, L., Zhao, J., Gurkar, A., Niedernhofer, L. J. & Robbins, P. D. Methods to quantify the NF-κB pathway during senescence. Methods Mol. Biol. 1896, 231–250 (2019).
Zhang, B. et al. KDM4 orchestrates epigenomic remodeling of senescent cells and potentiates the senescence-associated secretory phenotype. Nat. Aging 1, 454–472 (2021).
Karin, O., Agrawal, A., Porat, Z., Krizhanovsky, V. & Alon, U. Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nat. Commun. 10, 5495 (2019).
Gonzalez-Gualda, E. et al. Galacto-conjugation of navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19, e13142 (2020).
Guerrero, A. et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 1, 1074–1088 (2019).
Wong, C. H., Siah, K. W. & Lo, A. W. Estimation of clinical trial success rates and related parameters. Biostatistics 20, 273–286 (2019).
Newman, J. C. et al. Creating the next generation of translational geroscientists. J. Am. Geriatr. Soc. 67, 1934–1939 (2019).
Acknowledgements
This work was supported by the US National Institutes of Health (grants R37AG013925, R33AG061456, R01AG072301, R01AG061414, P01AG062413 and UH3AG056933), the Connor Fund, Robert J. and Theresa W. Ryan and the Noaber Foundation.
Author information
Authors and Affiliations
Contributions
J.L.K. conceptualized this Review. All authors contributed to the writing and approved the submitted version of the manuscript. S.C. created the figures using BioRender.com.
Corresponding author
Ethics declarations
Competing interests
T.T. and J.L.K. have a financial interest related to this research, including patents and pending patents covering senolytic drugs and their uses that are held by Mayo Clinic. S.C. has received royalties from Rejuveron Senescence Therapeutics. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with the Mayo Clinic’s conflict of interest policies.
Peer review
Peer review information
Nature Medicine thanks Masashi Narita and the other, anonymous, reviewer(s) for their contribution to the peer review. Primary Handling Editor: Karen O’Leary, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chaib, S., Tchkonia, T. & Kirkland, J.L. Cellular senescence and senolytics: the path to the clinic. Nat Med 28, 1556–1568 (2022). https://doi.org/10.1038/s41591-022-01923-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-022-01923-y
This article is cited by
-
NR2F2 alleviates pulmonary fibrosis by inhibition of epithelial cell senescence
Respiratory Research (2024)
-
Bazi Bushen ameliorates age-related energy metabolism dysregulation by targeting the IL-17/TNF inflammatory pathway associated with SASP
Chinese Medicine (2024)
-
Effects of high-intensity training on fatty infiltration in paraspinal muscles in elderly males with osteosarcopenia – the randomized controlled FrOST study
BMC Geriatrics (2024)
-
MicroRNA-377-3p exacerbates chronic obstructive pulmonary disease through suppressing ZFP36L1 expression and inducing lung fibroblast senescence
Respiratory Research (2024)
-
Emerging role of senescent microglia in brain aging-related neurodegenerative diseases
Translational Neurodegeneration (2024)
