
Abstract
Despite the success of antiretroviral therapy (ART) for people living with HIV, lifelong treatment is required and there is no cure. HIV can integrate in the host genome and persist for the life span of the infected cell. These latently infected cells are not recognized as foreign because they are largely transcriptionally silent, but contain replication-competent virus that drives resurgence of the infection once ART is stopped. With a combination of immune activators, neutralizing antibodies, and therapeutic vaccines, some nonhuman primate models have been cured, providing optimism for these approaches now being evaluated in human clinical trials. In vivo delivery of gene-editing tools to either target the virus, boost immunity or protect cells from infection, also holds promise for future HIV cure strategies. In this Review, we discuss advances related to HIV cure in the last 5 years, highlight remaining knowledge gaps and identify priority areas for research for the next 5 years.
Similar content being viewed by others

Main
Modern antiretroviral regimens can effectively block HIV replication in people with HIV for decades, but these therapies are not curative and must be taken for life. However, there is evidence that a cure can be achieved; initially, this came from a single case study (Timothy Brown, a man living with HIV who became widely known as the ‘Berlin patient’) following bone-marrow transplantation from a donor who was naturally resistant to HIV1. On the basis of this inspiring development and the recognition that not everyone can access and/or adhere indefinitely to antiretroviral therapy (ART), a global consensus emerged approximately 10 years ago that a curative intervention was a high priority for people with HIV and would be necessary to bring an end to the HIV pandemic. Since then, there has been a second case report of a cure following bone-marrow transplantation2 as well as evidence of persistence of only defective forms of the virus in certain patients3 and enhanced immune control of the virus by others after only a short time on ART4—further supporting the notion that a cure for HIV can be achieved.
An HIV cure includes both remission and eradication. Here, we define the term remission as durable control of virus in the absence of any ongoing ART. Eradication is the complete removal of intact and rebound-competent virus. The minimal and optimal criteria for an acceptable target product profile for an HIV cure, including the duration and level of virus control off ART, has recently been developed and published by the International AIDS Society (IAS), following wide consultation with multiple stakeholders5.
In 2011 and 2016, the IAS convened expert working groups to outline a strategy for developing an effective and scalable cure6,7. Since then, significant progress has been made, and the overall agenda has evolved. Here, we assembled a group of experts from academia, industry, and the community (Box 1) to evaluate recent progress and to outline cure-related research priorities for the next 5 years. The key recommendations for each component of the strategy are summarized in Box 2.
Understanding HIV reservoirs
A shared definition of the HIV reservoir is crucial for researchers, clinicians, and people living with HIV. Here, we use the term ‘HIV reservoir’ in the context of eradication or remission, as a representative term for all cells infected with replication-competent HIV in both the blood and different anatomical sites in individuals on ART—in other words, all potential sources of viral rebound in the context of a treatment interruption. Although the source of virus rebound is still not entirely understood, we now know that virus can persist in multiple forms, in multiple cells and in multiple sites.
Characterization of the complete HIV reservoir
HIV DNA can be detected in CD4+ T cells in blood and lymphoid tissue in nearly all people with HIV on ART. These viral genomes are mainly defective. Only a small proportion (less than 5%) appear to be intact and potentially replication-competent8. But the HIV reservoir goes beyond circulating CD4+ T cells; it also includes tissue-resident CD4+ T cells and cells of the monocyte/macrophage lineage, further complicating efforts to characterize and quantify it. In vitro, HIV preferentially integrates into transcriptionally active genes9; however, in people with HIV on ART, many proviruses (defined as virus that is integrated into the host genome), including intact ones, have been identified in genomic regions that are silent (known as ‘gene deserts’), which limits or precludes their reactivation3.
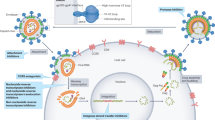
Our initial conception of the HIV reservoir as a static viral archive has given way to a more dynamic view in which, over time on ART, certain within-host HIV variants are gradually eliminated while others persist through various mechanisms, including clonal expansion of infected cells10,11,12,13,14,15. Sporadic infection of new cells during ART has been reported16, although there has been no convincing demonstration that viral sequences evolve during effective ART17, suggesting that the degree of virus spread is minimal. The sources of viral rebound following cessation of ART are incompletely defined. Multiple factors can contribute to viral replication following ART, including anatomical and microanatomical locations, the infected cell type, cellular phenotype, the nature of the provirus, the antigen specificity of the infected cell, the potential for transcriptional activity given the specific integration site, and/or distribution of antiretroviral drugs within tissues (Fig. 1).

The HIV reservoir can be defined across a number of dimensions, including: (1) anatomical and microanatomical locations, (2) cell type (for example, CD4+ T cell or macrophage), (3) cell functional profile (activated or resting; resistance to killing), (4) pool of proviruses with a particular functional profile (for example, interferon-alpha resistant) or (5) triggering event (for example, response to stimulation with a particular antigen), and (6) integration-site features of the rebounding virus.
We recommend prioritizing efforts to understand integration sites of the virus during long-term ART and to understand the inducibility of a provirus on the basis of its chromosomal context. In addition, large prospective studies incorporating analytical treatment interruptions (ATIs) are still needed to probe clinically relevant sources of viral rebound and to identify a biomarker that predicts this. A favorable cure intervention could either prolong the time to the point when virus is detectable (that is, rebound) in plasma or reduce the viral ‘set point’ (that is, post-treatment control).
One of the most daunting obstacles to designing more effective methods to target persistent HIV infection is the lack of biomarkers to unambiguously identify the cells that harbor the rebound-competent reservoir. Recent work has demonstrated that the viral reservoir is preferentially enriched in cells that express programmed death-1 (PD-1) and other immune checkpoint markers, activation markers such as HLA-DR, and chemokine receptors such as CCR6 and CXCR3, but there is no phenotypic marker specific for the reservoir18,19,20,21. Specific biomarkers of the reservoir are needed, particularly to assess the impact of cure interventions. Furthermore, understanding how HIV persists in specific tissue sites and relevant local cell populations, such as those in the brain, gastrointestinal tract liver, or genital tract, will be important, given that the mechanism for persistence in each site may be distinct, and therefore different approaches may be required to eliminate each of these reservoirs.
There is growing evidence that some defective proviruses can produce transcripts and proteins (including novel viral RNAs and chimeric viral proteins) that in turn can elicit immune responses and perhaps contribute to chronic inflammation22,23,24,25. This may be of high relevance to end organ complications, such as HIV-associated neurological disease26. If the production of RNA and proteins from these defective proviruses proves to have clinical relevance, then their removal may be necessary to ensure long-term health.
A major mechanism of HIV persistence is the proliferation of cells that were infected prior to ART, resulting in large clonal populations of infected cells that arise as a result of the site of HIV integration27,28, response to antigen29,30, or homeostatic drivers31. Characterization of these presumably physiological expansions might lead to the development of therapies aimed at interrupting proliferation of infected cells. It will be important to determine to what degree these expanded clones are transcriptionally active, whether they are an important of post-ART viral rebound, and whether they have some innate survival advantage that prevents the cells from being effectively cleared by the host.
Recent studies have provided some evidence for preferential survival of infected cells with proliferative advantages or with deeper viral latency. Prosurvival and immune-resistance profiles may be particularly important in infected cells that persist despite expression of viral RNA or proteins32,33,34. Opportunities likely exist for collaboration and cross-fertilization of concepts with the cancer field, where the clonal dynamics of tumors have been extensively studied in relation to prosurvival and immune-resistance advantages, such as the work being done on lung cancer through prospective genetic studies in TRACRx (https://clinicaltrials.gov/ct2/show/NCT01888601).
Biological sex can influence HIV pathogenesis, the immune response to HIV infection, and response to antiviral therapy35. Furthermore, in some but not all studies, women’s reservoirs have been shown to be less transcriptionally active and less inducible than those of men36,37,38,39,40. Sex, therefore, is a critical variable that should be considered as new therapies to target the reservoir are developed.
Quantification of the HIV reservoir
Significant progress toward a cure for HIV depends on having sensitive, specific, and quantitative measures of persistent virus that can be applied to various anatomical compartments41. Achieving this has been challenging, however, owing to the many sources and heterogeneous properties of persistent, replication-competent HIV. The reservoir can be quantified using assays that measure viral nucleic acid (total and integrated DNA, intact and defective DNA, or different forms of RNA), virus protein (p24), or viral inducibility (by measuring HIV RNA or virus replication following activation in vitro). Each approach has advantages and limitations, and assay outcomes may not always be interchangeable, comparable, or even correlated8.
Several groups have developed droplet digital PCR-based assays that discriminate genetically intact proviruses from a large background of defective proviruses, which are slightly less accurate but more high throughput than full genome sequencing42,43. The application of these assays to large clinical cohorts has demonstrated that there is a modest decrease in the frequency of cells with intact provirus over years on ART44,45,46. These assays have largely been optimized for subtype B virus, the major HIV subtype found in the United States and Europe. Yet there are over ten subtypes worldwide, some of which have evolved different mechanisms for immune evasion and persistence47. Pan-subtype-specific assays will need to be developed, and challenges related to cost and scalability remain. Research in this field should ideally culminate in harmonization across laboratories and crossvalidation of results. Future work will need to expand from quantification of virus in blood to quantification in tissue, particularly the more accessible tissues such as lymph nodes and gut mucosa.
Understanding the proviral landscape (defined as the degree of intactness, its transcriptional activity, and its location) is crucial, as these characteristics almost certainly influence the degree to which a provirus will rebound48. Over the last decade, several assays have been developed to analyze the exact location at which the virus integrates and whether the integrated virus is intact or defective. The ability to analyze single cells for integration site, viral sequence, and transcription is a major advance48; however, these assays are expensive and low throughput. Technological advances are required to apply this more broadly to clinical samples, including assessment of interventions that target the reservoir.
Cell-associated viral RNA (CA-RNA) provides a measure of the total transcriptional activity of proviruses within a given sample. Several assays have recently been developed that quantify different RNA species, including total, elongated, unspliced, polyadenylated, and multi-spliced RNA, and these stand to give higher-resolution insights into the impact of therapeutic intervention49. An important unmet need is to develop approaches to distinguish transcripts arising from defective versus intact proviruses. Another shortcoming hampering broad use of RNA assays is the fact that they are subtype-sensitive. Overall, our ability to study the biology of transcriptionally active proviruses and the role of transcriptional activity as a potential biomarker needs to be further explored.
Since HIV protein expression is also required for recognition by HIV-specific T cells and other immune-based therapies, measuring and characterizing viral proteins in cells and tissues is an important step to understanding HIV persistence and might prove to be a critical determinant for the efficacy of therapies that target the HIV-infected cells directly (for example, chimeric antigen receptor (CAR) T cells or broadly neutralizing antibodies). Quantification of the p24 protein with ultrasensitive enzyme-linked immunosorbent assays can determine the efficacy of therapies that target the reservoir directly. Ultrasensitive p24 assays have emerged as useful tools25, but drawbacks include low levels of sensitivity compared with nucleic acid detection, overestimation of the replication-competent reservoir, and the requirement for specialized instrumentation25,50. Detection of viral envelope protein (the target of many therapeutic interventions for an HIV cure) also remains a challenge. Future strategies should leverage advances in single-cell techniques and new approaches to imaging tissue using super-resolution or expansion microscopy, together with multi-omics approaches.
Substantial progress in other fields of medicine has been made in using advanced imaging techniques to quantify rare diseased cells in tissues. On the basis of some preliminary success in nonhuman primate models51, efforts to use radiolabeled HIV-specific tracers and sensitive imaging modalities (for example, positron emission tomography, PET) have been initiated52. Similar efforts aimed at characterizing sites of inflammation or expression of specific surface markers that are associated with HIV persistence should also be a priority.
Several studies have attempted to identify sources of rebound virus by probing phylogenetic linkages with the proviral sequences present in various anatomical and cellular compartments. Success has been limited, however, in part owing to the challenging nature of obtaining full-length sequences from the limited number of infected cells in blood or tissue, as well as from plasma with low level viremia53. Strategies that can enhance enrichment of infected cells and/or depth of viral sequencing together with high-throughput low-cost single-cell analyses are likely to advance the field. As the RNA in circulating virions is a well-accepted surrogate marker for untreated HIV disease, this measurement could be an effective tool to characterize the rebound-competent population of HIV-infected cells.
Currently, any impact of a therapeutic intervention on the viral reservoir can only be determined with an ATI. A tool for very early detection of viral rebound post-ART using a nonvirological marker—such as measures of the innate immune response54—could be very valuable. In addition, better ways to monitor viral load that do not require frequent healthcare appointments will be needed5. This should include the development of home-based tests that may not necessarily require high sensitivity as long as testing is performed frequently55. Finally, emerging evidence suggests that virus replication during an ATI may be associated with some long-term adverse events56, so careful follow up of participants in ATI studies will be necessary.
Mechanisms and models of virus control
Natural control in people living with HIV
Individuals who naturally control HIV in the absence of any therapy and can maintain a viral load of <50 copies/ml (known as ‘elite’ controllers) have been the focus of intense investigation for years. Research in this area is increasingly focused on those controllers who exhibit remarkably stringent control (‘exceptional’ controllers)57,58, some of whom might be considered true cures48,59, and those who became controllers after ART interruption (post-treatment controllers)60,61. In exceptional controllers, the frequency of infected cells is extremely low, often below the limit of detection of most standard assays for HIV DNA57,59, there is no intact virus48 and the site of HIV integration may be distinct48; an agreed definition for an exceptional controller is needed.
Virus-specific CD8+ T cells targeting particularly vulnerable or conserved epitopes are generally recognized as the key mediator of elite control; such cells are rare in post-treatment controllers and have not yet been characterized in exceptional controllers4,62. Further characterization of the various controller phenotypes (elite, exceptional, post-treatment) should remain a priority; the identification of unique and potentially informative phenotypes should also be pursued, including individuals on ART who have very small reservoirs62. Functional multi-omics studies and emerging single-cell technologies should help to determine the mechanisms involved in exceptional, elite, and post-treatment control. Better animal models of exceptional and post-treatment control would greatly enhance the field, giving access to tissue and the opportunity for longitudinal assessment of virus control63.
Virus elimination and control will likely require a coordinated immune response involving more than just T cells. Recent data suggest that autologous antibodies targeting archived viruses as well as interferon sensitivity might influence which virus populations emerge post-ART54,64,65. Studies in simian immunodeficiency virus (SIV)-infected nonhuman primates that naturally control infection have provided indirect evidence that natural killer (NK) cells might be able to effectively control virus in tissues66. Better insights into the role of antibodies, natural killer cells, and innate immunity in post-treatment and/or post-intervention control are needed.
The interplay between the virus and immune system during acute infection or immediately after the interruption of ART is largely unknown, at least in humans. During acute infection, those destined to become controllers typically have an initial period of poorly controlled viremia61,67. For post-treatment controllers, virus control is often achieved more rapidly after cessation of ART than after primary infection61,68. We need to understand the viral dynamics associated with eventual post-ART control/remission, as this will inform how a treatment interruption should be conducted. It is likely that biomarkers other than the plasma HIV RNA level might allow for the development of safer and more cost-effective strategies for interrupting ART.
Animal models of control
The role of humanized mouse models in cure research is still evolving. Recent studies showing similar effects of latency-reversing strategies in mice and the less scalable nonhuman primate model are encouraging69,70. Given that access to nonhuman primates for cure studies will likely remain a barrier, ongoing optimization, standardization, and validation of mouse models should be prioritized.
An important discrepancy in translating cure-related findings from SIV-infected nonhuman primates to people with HIV lies in the duration of ART. Although effective ART regimens with integrase inhibitors have been optimized in nonhuman primates, high costs, and treatment-related toxicities necessitate relatively shorter study durations (less than 1–2 years of ART). One possible solution would be for primate research centers to maintain colonies of SIV-infected nonhuman primates receiving very-long-term ART to be directly assigned for studies.
There is ongoing debate about the most appropriate virus to be used in cure-related studies in nonhuman primates. Investigations utilizing broadly neutralizing antibodies or select vaccines directed against the HIV-1 envelope necessitate infection with a virus that expresses HIV envelope proteins (simian-human immunodeficiency virus, SHIV). However, SHIV infections with some strains are characterized by post-treatment control in the absence of any intervention71, while others can induce significant disease progression72. Therefore, the specific strain used can limit the generalizability of the model. Although SIV infection of nonhuman primates can cause more significant disease progression than HIV infection of people, early ART for SIV infection can limit rapid disease progression and is therefore a useful model for cure studies73. Developing immunotherapies that target the SIV envelope in addition to SHIV should also be pursued.
A major recent advance has been the development of genetically barcoded SIVmac239 strains74. Because the barcode ‘tags’ are easily quantified and also passed on to progeny virus, this model allows for tracking of clonal dynamics, providing more precise insights into how interventions affect seeding of the reservoir, viral reactivation during ART, or viral recrudescence after ART interruption.
Therapeutic interventions
Targeting the provirus
Since the discovery that HIV can establish a latent infection with minimal HIV transcription, a range of approaches has emerged that specifically target latently infected cells. These include pharmacological modulation of epigenetic or signaling pathways involved in HIV transcription to reactivate latent HIV such that the cells can be targeted and eliminated (‘shock and kill’) or to permanently silence HIV transcription (‘block and lock’)75,76,77. Recent reports have demonstrated that HIV latency is heterogeneous and that latency reactivation is stochastic, implying that a combination of agents targeting various pathways controlling HIV transcription may be necessary to achieve either robust silencing or latency reversal49,78,79,80.
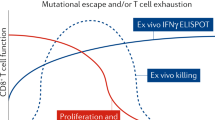
A clear limitation of the ‘shock and kill’ approach comes from the discovery that only a fraction of proviruses is intact and among these, only some are inducible by a potent stimulus such as T cell stimulation, let alone by far less potent latency-reversing agents (LRAs)8,81,82,83. Furthermore, cells containing reactivated latent HIV may also be relatively resistant to killing by cytotoxic T cells84. Complicating the situation even more, CD8+ T cells appear to suppress HIV transcription and can blunt the effect of LRAs70.
Although LRAs tested in humans can induce HIV RNA expression and virion production in vivo, they have failed to reduce the size of the reservoir, even when combined with immunotherapeutic strategies designed to enhance clearance of infected cells85,86,87,88,89,90. This could be due to poor antigen induction by LRAs or insufficient clearance of these targets by immunotherapies (Fig. 2). Furthermore, many of the tested LRAs have off-target effects. Newer approaches for delivery of LRAs to reduce toxicity, enhance potency, and improve targeting, potentially leveraging advances in nanomedicine, should be explored. Greater potency could potentially be achieved using LRAs in combination, however, care is needed in these clinical trials, given that unexpected toxicities can emerge—as was recently demonstrated in the evaluation of high-dose disulfiram and vorinostat91. Finally, LRAs will likely need to be partnered with therapies that enhance the clearance of cells expressing viral proteins, such as immune-enhancing strategies or proapoptotic drugs92.

Reversing latency is an important component of revealing HIV-infected cells, allowing for conversion of a latently infected to a productively infected cell. a, Currently available LRAs reverse latency in only a subset of infected cells, and, when used alone, do not sufficiently eliminate these. b, Enhancing the efficacy of an LRA can be achieved with increased potency, targeted delivery or through using combinations of LRAs. c, Ultimately, depletion of the reservoir will require combining an LRA with other interventions, such as immunotherapy or a proapoptotic drug.
Permanently silencing the HIV promoter by suppressing factors that promote HIV transcription has also emerged as a strategy to target the provirus. The concept is to therapeutically drive HIV into a permanently silenced epigenetic state that resists reactivation (‘deep latency’). The Tat inhibitor didehydro-cortistatin A (dCA) blocks HIV reactivation from human CD4+ T cells in vitro through epigenetic repression; treatment with dCA in ART-suppressed humanized mouse latency models induces a measurable delay in virus rebound76,93. Gene therapy can also play an important part in permanent silencing of the provirus using short interfering RNA or other modalities94. Thus far, these approaches have yet to be successfully translated into human trials.
Further exploration of the therapeutic potential of permanently silencing the reservoir (‘block and lock’), presumably as part of a combinatorial cure approach, is a high research priority. Some pathways that might be targeted include mTOR, HSF1, and others95,96,97. Efforts to screen for drugs that suppress HIV transcription are encouraged,96,98,99 with the goal to rapidly move into preclinical and clinical studies100,101.
With a recent report indicating that the HIV reservoir is stabilized at the start of ART initiation, efforts should be devised to inhibit this stabilizing effect and/or to enhance reservoir turnover during ART, where such interventions are ideally delivered at ART initiation102,103,104.
Most methods to target the provirus have been developed using subtype B. Thus, while conserved mechanisms govern latency across the different virus subtypes, differences at the level of the promoter may impact responsiveness to various stimuli. Therapies targeting the provirus should be evaluated across multiple HIV subtypes including recombinants.
Targeting the immune system
There is a robust and growing toolbox of immune therapies that might be advanced to proof-of-concept testing. Arguably, the most impactful innovation to date is the isolation and development of broadly neutralizing antibodies for clinical use, but advances have also been made in the development of therapeutic vaccines, vaccine adjuvants, and other immunotherapies. When used in combination in nonhuman primates, these immune therapies have resulted in sustained post-ART control71,105. When used alone, most of these approaches have had limited effectiveness in people, although some promising results are emerging88,106,107. Combination clinical trials have recently started and are ongoing. Although the combination of either vorinostat or romidepsin (HDAC inhibitors that can increase viral transcription through epigenetic modification) together with different HIV vaccines showed no or minimal reduction in the HIV reservoir107,108,109, results from other studies including a combination of Toll-like receptor agonists, LRAs and broadly neutralizing antibodies are eagerly awaited (NCT03837756; NCT04319367; NCT03041012).
As it may be challenging to reactivate and eliminate all latently infected cells, or to induce deep irreversible latency in all cells, it seems unlikely that these approaches will be curative by themselves. By reducing the reservoir, however, they might make strategies aimed at controlling the virus long term post-ART more effective. This overall approach of 'reduce and control’ is supported by observations in elite and post-treatment controllers, and theoretical modeling110. Multiple approaches that might result in control of a small reservoir are being developed. Assessment of therapeutic vaccines including live vector vaccines such as adenovirus 26, modified vaccine Ankara, and also a cytomegalovirus in nonhuman primate models have been particularly promising, with a subset of animals achieving eradication of virus71,111. Such studies have not yet been performed in people. Research to develop and test novel immunogen and vaccine designs with broad, potent and durable immunity should be prioritized. Given the recognition that autologous neutralizing antibodies might contribute to reservoir control64, novel vaccine approaches aimed at the induction of broadly neutralizing antibodies—including germline targeting112—should also be prioritized.
Immune stimulators, immunomodulators, and novel immunotherapies (such as cytokine formulations, Toll-like receptor agonists, immune checkpoint inhibitors or agonists, and novel vaccine adjuvants), used alone or more likely in combination with other approaches, hold promise but have undergone relatively limited testing in HIV-cure studies in people so far106,113,114,115.
With the exception of a few anecdotal cases116, immunotherapy in people with HIV has yet to recapitulate the promising advances made in nonhuman primates. Combination of various therapies will almost certainly be needed (Fig. 3). Conducting such studies is feasible108,117; it is expected that initial clinical research will be intensive in nature and designed to identify strategies that might then be tested in well-powered, controlled clinical trials. Defining the mechanisms and potential biomarkers associated with remissions/cures in the preclinical and clinical setting should remain a priority. Determining which combinations to study, and how to define the optimal doses and strategies, poses a significant challenge from a methodological and regulatory perspective. As immunotherapies for HIV move into the clinic, careful attention will have to be paid to immune-related adverse events, including cytokine-release syndrome and autoimmunity.

Strategies that will enhance immune-mediated clearance of latently infected cells include early initiation of ART and the administration of combined interventions at the time of suppressive ART (colored arrows) or during the treatment interruption phase, which will allow for increased antigen presentation. Given that there is no biomarker that can predict viral rebound, analytical treatment interruptions are used to determine whether the intervention has had a clinically meaningful impact. The overarching goal is to either delay viral rebound by at least months or years or reduce the set point of virus replication (that is, the stable level of viral load that the body settles at), preferably to a level of <200 copies/ml. The dashed colored lines represent different potential favorable outcomes from a cure intervention. bNAbs, broadly neutralizing antibodies; LRA, latency reversing agent; TLR, Toll-like receptor.
Cell and gene therapy
Cell and gene therapy clinical trials for people with HIV, although safe so far, have been small in scale and with no clear demonstrations of efficacy. The interest in gene therapy for an HIV cure was inspired by the elimination of intact virus in Timothy Brown (also known as the Berlin patient) and Adam Casteljo (also known as the London patient), who both received stem-cell transplants from a CCR5-negative donor1,2 to treat their underlying malignancies. CCR5 is a co-receptor that is needed by most strains of HIV to enter a cell; a reduction in the size of the reservoir has also been reported following stem-cell transplantation to people with HIV from donors who are CCR5-positive118,119, but the HIV reservoir can’t be completely eliminated, irrespective of the CCR5 status of the donor. In the case of CCR5-negative stem-cell transplantation, the absence of CCR5 in the donor cells is thought to protect the newly transplanted cells from infection, at least with CCR5-dependent HIV strains. Interestingly, in both cases of cure following stem-cell transplantation of CCR5-negative cells, defective virus has been detected, but not intact or replication-competent virus120,121. These reports have prompted researchers to evaluate CCR5-targeted gene editing as a potentially safer path to cure in people living with HIV on ART, given the high mortality rate and significant morbidity associated with stem-cell transplantation. Timothy Brown unfortunately died in early 2020 owing to recurrence of his leukemia, but remained HIV-free until his death.
Ex vivo gene editing of CCR5 using zinc finger nucleases and re-infusion of CCR5-modified T cells has not yet prevented viral rebound following ATI122,123, possibly because insufficient cell numbers were engineered and/or engrafted with first-generation editing tools and cell culture protocols and/or because CCR5 disruption alone cannot shift the balance in favor of post-treatment control in the presence of persistently infected cells. More recently, gene therapies have shifted to creating effectors, including chimeric antigen receptor (CAR) T cells, which can recognize and eliminate HIV-infected cells (Fig. 4). Other approaches include the use of novel delivery systems to deliver genes to local tissues, resulting in the sustained production of systemically acting antivirals such as broadly neutralizing antibodies124,125 and CD4 mimetics126. Finally, attempts are being made to directly target integrated proviruses with technologies such as CRISPR–Cas9 and recombinases127,128. This approach remains conceptually challenging in view of the disparate locations of latently infected cells, the absence of specific markers to target delivery, the heterogeneity of proviral sequences (the majority of which are defective), and the risk of off-target effects.

Examples of ex vivo (left) and in vivo (right) gene therapy approaches that have been tested in people with HIV on ART. Ex vivo strategies include gene editing to either delete or inactivate CCR5 or HIV provirus in CD4+-enriched T cells using gene-editing tools such as zinc finger nucelases (ZFN) or CRISPR–Cas9. Alternatively, autologous T cells can be modified to express a CAR that can recognize HIV envelope, and this can then be reinfused into the participant. In vivo strategies, on the other hand, do not require external manipulation of cells; nanoparticles or viral vectors (such as adeno-associated virus (AAV)), which encapsulate mRNA or DNA, respectively, for the relevant gene to be expressed are administered directly to the patient. These approaches have recently been successful using lipid nanoparticles that contain mRNA encoding CRISPR–Cas9135 or for expression of anti-HIV broadly neutralizing antibodies such as PG9 or VRC07 (ref. 125). PBMCs, peripheral blood mononuclear cells; PLWH, person living with HIV.
Many emerging cell and gene therapies are designed to target viral proteins/epitopes that are expressed in abundance on the surface of tumor cells, for example CD19 for the treatment of lymphoma129. Various forms of the HIV viral envelope protein (gp120 trimers and monomers, gp41) are expressed on the surface of infected cells, while multiple peptides are presented via HLA molecules. These antigens are expressed at levels well below that of many cancer antigens now being successfully targeted in the clinic. Cell-based therapies such as CAR T cells, once infused into the patient, will only persist and differentiate if there is sufficient antigenic exposure; however, the levels of antigen during ART may be too low130. Removal of ART after infusion of CAR T cells (or similar products) could be used to expand these cells in vivo, or more potent latency-reversing agents could be used to enhance envelope protein expression. In addition, novel adjuvants could expand CAR T cells even when the antigen burden is low, as was recently demonstrated in the nonhuman primate model131.
The challenges here are primarily those of delivery to relevant cells. In addition to developing methods to target specific cells, which are common problems faced by all potential in vivo gene therapies, targeting latent proviruses also presents the problem of a lack of robust cell surface markers to identify cells harboring such proviruses. Progress in both of these areas will be needed to develop strategies to deliver gene-editing reagents to latently infected cells. Some promising in vivo delivery strategies for CRISPR–Cas9 have included adeno-associated virus to target the SIV virus in nonhuman primates on ART127, as well as using engineered CD4+ cell-homing messenger RNA (mRNA)-containing lipid nanoparticles in mouse models of HIV infection132.
Long-term in vivo secretion of antibodies or antibody-like molecules can be achieved following gene therapy vector delivery of antibody cassettes to tissues such as muscle and liver, where enhanced production of antibodies is needed, rather than specific delivery to infected CD4+ T cells. This can be achieved through direct intramuscular injection leading to uptake in the muscle or, alternatively, intravenous injection, which will allow for uptake in the liver. Ectopic expression of these antibodies in liver or muscle cells fails to recapitulate aspects of natural antibody production, such as responsiveness to antigen and ongoing somatic hypermutation. Therefore, editing the B cell Ig locus itself to express antibodies presents an alternative and attractive gene-editing strategy133,134.
Sustained production of these antivirals could result in sustained (perhaps lifelong) control of the virus. Many barriers to success exist. Antidrug antibodies that target and clear the vectors often form rapidly125, limiting the ability to deliver multiple doses. Advances in mRNA encapsulation within lipid nanoparticles may potentially revolutionize delivery of gene therapy, allowing for delivery of mRNA encoding CRISPR–Cas9 and related guide RNAs in vivo, as has recently been successfully demonstrated in the treatment of transthyretin amyloidosis135. Also, antibodies targeting multiple antigens will likely need to be produced at high levels to prevent virus replication and escape.
Advances in T cell manufacturing are expected, driven by cancer CAR T cell therapies, which will also benefit HIV therapies. Similarly, advances occurring in gene therapy treatments for genetic diseases, such as hemoglobinopathies, are catalyzing safer and nongenotoxic conditioning for HSPC transplants, for example based on drug–antibody conjugates. Practicality will also be enhanced by moving toward using allogeneic off-the-shelf products.
Gene and cell therapies now require a shift towards a practical focus, identifying ways to expand use, reduce costs, and allow deployment in resource-limited settings. This could be achieved through abbreviated ex vivo cell manufacturing, including automated closed-system devices (‘gene therapy in a box’), to produce product in a place-of-care setting136. While still in the early stages of development, in vivo gene therapy also presents exciting possibilities to significantly expand access by eliminating the need for external manipulation of cells and associated technological requirements.
Pediatric remission and cure
The unique context of perinatal HIV infection necessitates pediatric-specific strategies to achieve ART-free remission in children. The case of the Mississippi child, who started therapy ~30 hours after birth and achieved remission off ART for 27 months before virus rebounded137,138, raised the possibility that remission for children can be attained. Subsequent reports of early-treated pediatric cases with long-term (>12 years) virological control off ART have provided examples of post-treatment control in children139,140.
The nature of the reservoir in children is unique from that in adults. For example, naive CD4+ T cells are a more important reservoir for the virus in children141,142. Further development of infant nonhuman primate models for evaluating ART and cure strategies will contribute to our understanding of the HIV reservoir and how to target it in the unique setting of infancy and immune development, but an understanding of the limitations of this model is also crucially important141,142,143,144,145.
Many of the recent advances in understanding HIV persistence during ART in adults, including frequency and transcriptional activity of intact virus, clonal expansion, sites of proviral integration, and inducibility, need to be applied to studies of children. Optimizing methods that can be adapted to small blood volumes are also needed.
In the context of childhood infection, clarity is needed on how latency is established in naive T cells, susceptibility of these cells to latency reversal, propensity for T cells to clonally expand, and the relative contribution of clonally expanded cells to viral rebound following cessation of ART. It is still unclear whether integration sites and reactivation potential are different in children, and whether these change with age. Given that initial studies suggest a less-inducible reservoir in cases of perinatal infection146, it is especially important to determine how to maximize latency reversal in children. The optimal timing of these interventions (for example, at the time of early ART initiation) could potentially limit the pool of infected cells that persist on ART; such approaches can be explored in a nonhuman primate model.
As in adults, better tools are needed to assess the impact of cure interventions in children, including quantification of HIV persistence and in-depth cellular immune profiling. There is a particular need for noninvasive tools, such as total body imaging, to assess central nervous system and other tissue-based reservoirs. It will also be important to identify biomarkers for post-treatment control, including the degree of reduction or alteration in the composition of the latent reservoir that may be predictive of pediatric remission or cure147. Finally, preclinical studies in infant nonhuman primates that test new interventions to reduce or eliminate persistent HIV and/or induce viral remission after ART interruption are needed to inform the development of HIV remission and cure intervention strategies. Early therapy alone is insufficient to reliably achieve a cure or long-term remission in children. Novel approaches, including earlier administration and use of more potent antiretroviral drugs, therapeutic vaccines, or other immunotherapeutics, such as broadly neutralizing antibodies and/or innate-immune-enhancing agents, will be necessary.
Social, behavioral, and ethical aspects of cure
Research directed toward an HIV cure intertwines critical social, behavioral, and ethical aspects that must be incorporated in the scientific agenda. This research takes place within particular social contexts and communities that shape its permissibility and appropriateness. Accordingly, affected communities must be meaningfully engaged throughout the research process; social and behavioral factors must be interrogated and taken into account because they affect research feasibility, community support for the research, and the well-being of participants and other stakeholders. Research must also address the many ethical issues associated with developing a therapy, particularly since viable options for treatment are already available. Sufficient funding for research toward social, behavioral, and ethical aspects of a cure and for community involvement is therefore essential.
Substantial progress using more conceptual and normative approaches has also been made regarding the ethical issues associated with the interruption of ART148,149. Similarly, there has been attention focused on acceptable risk thresholds for research150. Finally, given the important role of treatment as prevention, efforts have focused on the ethics of partner-protection measures151.
Community engagement in HIV cure research is still suboptimal in many settings, being mostly been limited to advisory boards typically comprised of scientifically literate individuals. Capacity to discuss HIV cure research and to evaluate its potential implications for local and global communities must be built within diverse community groups. Communities should be empowered and supported through education and engagement at all levels of the research process to help shape the HIV cure research agenda and allow for potential study participants to have a voice in trial design152.
Since HIV cure research is highly complex and nuanced, there is also a need to ensure understanding of it among other key stakeholders, including Institutional Review Boards (IRBs) and clinicians. For example, IRBs need to appreciate the implications of ATIs for partners who they may not see as within their remit, and clinicians need to understand the rationale for ATIs in the research setting.
Attention must focus on broad representation (for example, age, race and ethnicity, gender and sexuality, geographic location, risk behaviors) in research. Diversity in participation is essential during the development of interventions aimed at complete HIV elimination or durable ART-free control. This necessitates research directed at understanding the reasons for under-representation of certain groups of people in HIV cure research. For example, cisgender and transgender women, as well as individuals of some racial and ethnic backgrounds, are less likely to participate in HIV-cure-focused clinical trials153. This highlights the need for more nuanced and theoretically engaged research to understand how gender, race, and other characteristics shape engagement with HIV cure research154. At the same time, legal and social considerations unique to each context must be identified and addressed. For example, local laws, stigma, and access to healthcare affect research involving the interruption of ART.
There is also a need to better define ethical considerations involved in the selection of populations of interest in which promising cure strategies will be tested. For example, should priority be given to testing new interventions in individuals who initiated treatment during acute infection over those who began treatment during chronic infection? What are the best means to identify and manage the ethical considerations in pediatric HIV cure research? In addition, what measures ought to be taken to ensure that recruitment is not skewed toward people with HIV in resource-limited settings? Further, ethical questions of equity and justice related to the distribution of safe and effective cure interventions must consider acceptability, scalability, and cost-effectiveness. The COVID-19 pandemic has raised unique considerations for research participants, staff, and communities155. Given the rapidly changing nature of the pandemic and the availability of COVID-19 vaccines and other treatments, there is a need to continually revise and assess the safety and feasibility of HIV cure research efforts.
During the study design phase, early engagement is needed in communities where research is being considered in order to determine the nature and acceptability of research-related risks. Similarly, stakeholder perceptions should be elicited to guide the development of target product profiles (the minimal and optimal characteristics of a new therapeutic intervention), as recently done for an HIV cure5. In especially complex clinical studies, formative research should be used to help develop a robust, informed consent process. Furthermore, nested social and behavioral research (basic, elemental, supportive, integrative) is needed to enhance understanding of the actual experiences of trial participants as well as of sexual partners of participants. These data will help provide a check on current practices, as well as provide a foundation for future efforts aimed at improving them.
Conclusion
Several of the key topics addressed in the previous sections are prerequisites for the development of successful cure strategies and interventions. To date, most HIV cure research has been restricted to high-income countries with relatively low HIV burden and has most often engaged men who have sex with men. HIV strains are genetically and biologically diverse, and host mechanisms of antiviral immunity required for durable control may differ by sex, geography, and ethnicity. Basic discovery research and clinical trials in resource-limited settings must be strengthened and will require enabling infrastructure development and capacity building.
In the next decade, we expect to see a greater understanding of HIV reservoirs, an increasing number of clinical trials and hopefully reports of individuals who achieved long-term remission with less intensive and more widely applicable strategies. On the basis of the current understanding and lessons from ART, it is likely that combinations of these approaches may be the first approach to be implemented. Inclusion of knowledge from fields such as oncology and COVID-19 could also greatly facilitate progress. Finally, open and responsible communication about trials and realistic expectations will remain important. Although safety is the highest priority, with increasing number of clinical trials, there is an increase in the possibility of adverse events which will need to be appropriately managed while allowing the field to advance.
This global scientific strategy, in combination with the recently developed target product profile5, will assist with guiding the field toward a widely applicable, acceptable, and affordable cure. The establishment of the HIV Cure Africa Acceleration Partnership152 will hopefully enable broader engagement and facilitate rapid implementation of any successes into low- and middle-income settings. Fortunately, the resources for such work remain available, and the field is highly committed to making the long-term commitments necessary to develop an effective and scalable remission or cure strategy.
References
Hutter, G. et al. Long-term control of HIV by CCR5 ∆32/∆32 stem-cell transplantation. N. Engl. J. Med. 360, 692–698 (2009).
Gupta, R. K. et al. HIV-1 remission following CCR5∆32/∆32 haematopoietic stem-cell transplantation. Nature 568, 244–248 (2019).
Jiang, C. et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 585, 261–267 (2020).
Saez-Cirion, A. et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 9, e1003211 (2013).
Lewin, S. R. et al. Multi-stakeholder consensus on a target product profile for an HIV cure. Lancet HIV 8, e42–e50 (2021).
International, A. S. S. W. G. O. H. I. V. C. et al. Towards an HIV cure: a global scientific strategy. Nat. Rev. Immunol. 12, 607–614 (2012).
Deeks, S. G. et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat. Med. 22, 839–850 (2016).
Eriksson, S. et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 9, e1003174 (2013).
Schaller, T. et al. HIV-1 capsid–cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439 (2011).
Cohn, L. B. et al. HIV-1 integration landscape during latent and active infection. Cell 160, 420–432 (2015).
Lee, G. Q. et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Investig. 127, 2689–2696 (2017).
Simonetti, F. R. et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl Acad. Sci. USA 113, 1883–1888 (2016).
Lorenzi, J. C. et al. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proc. Natl Acad. Sci. USA 113, E7908–E7916 (2016).
Bui, J. K. et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 13, e1006283 (2017).
Halvas, E. K. et al. HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J. Clin. Invest. 130, 5847–5857 (2020).
Fletcher, C. V. et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl Acad. Sci. USA 111, 2307–2312 (2014).
McManus, W. R. et al. HIV-1 in lymph nodes is maintained by cellular proliferation during antiretroviral therapy. J. Clin. Invest. 129, 4629–4642 (2019).
Neidleman, J. et al. Phenotypic analysis of the unstimulated in vivo HIV CD4 T cell reservoir. eLife 9, e60933 (2020).
Gosselin, A. et al. HIV persists in CCR6+CD4+ T cells from colon and blood during antiretroviral therapy. AIDS 31, 35–48 (2017).
Fromentin, R. et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog. 12, e1005761 (2016).
Anderson, J. L. et al. Human immunodeficiency virus (HIV)-Infected CCR6+ rectal CD4+ T cells and HIV persistence on antiretroviral therapy. J. Infect. Dis. 221, 744–755 (2020).
Imamichi, H. et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl Acad. Sci. USA 113, 8783–8788 (2016).
Imamichi, H. et al. Defective HIV-1 proviruses produce viral proteins. Proc. Natl Acad. Sci. USA 117, 3704–3710 (2020).
Pollack, R. A. et al. Defective HIV-1 proviruses are expressed and can be recognized by cytotoxic T lymphocytes, which shape the proviral landscape. Cell Host Microbe 21, 494–506(2017).
Wu, G. et al. Gag p24 is a marker of HIV expression in tissues and correlates with immune response. J. Infect. Dis. 224, 1593–1598 (2021).
Spudich, S. et al. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J. Clin. Invest. 129, 3339–3346 (2019).
Maldarelli, F. et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183 (2014).
Wagner, T. A. et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345, 570–573 (2014).
Mendoza, P. et al. Antigen-responsive CD4+ T cell clones contribute to the HIV-1 latent reservoir. J. Exp. Med. 217, e20200051 (2020).
Simonetti, F. R. et al. Antigen-driven clonal selection shapes the persistence of HIV-1-infected CD4+ T cells in vivo. J. Clin. Invest. 131, 145254 (2021).
Chomont, N. et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15, 893–900 (2009).
Ren, Y. et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J. Clin. Investig. 130, 2542–2559 (2020).
Kuo, H. H. et al. Anti-apoptotic protein BIRC5 maintains survival of HIV-1-infected CD4+ T cells. Immunity 48, 1183–1194(2018).
Cummins, N. W. et al. Maintenance of the HIV reservoir is antagonized by selective BCL2 inhibition. J. Virol. 91, e00012-17 (2017).
Scully, E. P. Sex differences in HIV infection. Curr. HIV/AIDS Rep. 15, 136–146 (2018).
Das, B. et al. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specific restrictions on the latent reservoir. Proc. Natl Acad. Sci. USA 115, E7795–E7804 (2018).
Prodger, J. L. et al. Reduced HIV-1 latent reservoir outgrowth and distinct immune correlates among women in Rakai, Uganda. JCI Insight 5, e139287 (2020).
Falcinelli, S. D. et al. Impact of biological sex on immune activation and frequency of the latent HIV reservoir during suppressive antiretroviral therapy. J. Infect. Dis. 222, 1843–1852 (2020).
Scully, E. P. et al. Sex-based differences in human immunodeficiency virus type 1 reservoir activity and residual immune activation. J. Infect. Dis. 219, 1084–1094 (2019).
Szotek, E. L., Narasipura, S. D. & Al-Harthi, L. 17β-Estradiol inhibits HIV-1 by inducing a complex formation between β-catenin and estrogen receptor alpha on the HIV promoter to suppress HIV transcription. Virology 443, 375–383 (2013).
Abdel-Mohsen, M. et al. Recommendations for measuring HIV reservoir size in cure-directed clinical trials. Nat. Med. 26, 1339–1350 (2020).
Gaebler, C. et al. Sequence evaluation and comparative analysis of novel assays for intact proviral HIV-1 DNA. J. Virol. 95, e01986-20 (2021).
Bruner, K. M. et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 22, 1043–1049 (2016).
Peluso, M. J. et al. Differential decay of intact and defective proviral DNA in HIV-1-infected individuals on suppressive antiretroviral therapy. JCI Insight 5, 132997 (2020).
Gandhi, R. T. et al. Selective decay of intact HIV-1 proviral DNA on antiretroviral therapy. J. Infect. Dis. 223, 225–233 (2020).
Anderson, E. M. et al. Dynamic shifts in the HIV proviral landscape during long term combination antiretroviral therapy: implications for persistence and control of HIV infections. Viruses 12, E136 (2020).
Sarabia, I. & Bosque, A. HIV-1 latency and latency reversal: does subtype matter? Viruses 11, 1104 (2019).
Jiang, C. et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 585, 261–267 (2020).
Yukl, S. A. et al. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 10, eaap9927 (2018).
Pardons, M. et al. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 15, e1007619 (2019).
Santangelo, P. J. et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Nat. Methods 12, 427–432 (2015).
McMahon, J. H. et al. A clinical trial of non-invasive imaging with an anti-HIV antibody labelled with copper-64 in people living with HIV and uninfected controls. EBioMedicine 65, 103252 (2021).
De Scheerder, M. A. et al. HIV rebound is predominantly fueled by genetically identical viral expansions from diverse reservoirs. Cell Host Microbe 26, 347–358(2019).
Mitchell, J. L. et al. Plasmacytoid dendritic cells sense HIV replication before detectable viremia following treatment interruption. J. Clin. Invest. 130, 2845–2858 (2020).
Draz, M. S. et al. DNA engineered micromotors powered by metal nanoparticles for motion based cellphone diagnostics. Nat. Commun. 9, 4282 (2018).
Richart, V. et al. High rate of long-term clinical events after ART resumption in HIV-positive patients exposed to antiretroviral therapy interruption. AIDS https://doi.org/10.1097/QAD.0000000000003058 (2021).
Mendoza, D. et al. Comprehensive analysis of unique cases with extraordinary control over HIV replication. Blood 119, 4645–4655 (2012).
Canoui, E. et al. A subset of extreme human immunodeficiency virus (HIV) controllers is characterized by a small HIV blood reservoir and a weak T-cell activation level. Open Forum Infect. Dis. 4, ofx064 (2017).
Casado, C. et al. Permanent control of HIV-1 pathogenesis in exceptional elite controllers: a model of spontaneous cure. Sci. Rep. 10, 1902 (2020).
Saez-Cirion, A. et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 9, e1003211 (2013).
Namazi, G. et al. The Control of HIV After Antiretroviral Medication Pause (CHAMP) Study: posttreatment controllers identified from 14 clinical studies. J. Infect. Dis. 218, 1954–1963 (2018).
Galvez, C. et al. Extremely low viral reservoir in treated chronically HIV-1-infected individuals. EBioMedicine 57, 102830 (2020).
Passaes, C. et al. Optimal maturation of the SIV-specific CD8+ T cell response after primary infection is associated with natural control of SIV: ANRS SIC Study. Cell Rep. 32, 108174 (2020).
Bertagnolli, L. N. et al. Autologous IgG antibodies block outgrowth of a substantial but variable fraction of viruses in the latent reservoir for HIV-1. Proc. Natl Acad. Sci. USA 117, 32066–32077 (2020).
Gondim, M. V. P. et al. Heightened resistance to host type 1 interferons characterizes HIV-1 at transmission and after antiretroviral therapy interruption. Sci. Transl. Med. 13, eabd8179 (2021).
Huot, N. et al. Natural killer cells migrate into and control simian immunodeficiency virus replication in lymph node follicles in African green monkeys. Nat. Med. 23, 1277–1286 (2017).
Madec, Y. et al. Natural history of HIV-control since seroconversion. AIDS 27, 2451–2460 (2013).
Chun, T. W. et al. Effect of antiretroviral therapy on HIV reservoirs in elite controllers. J. Infect. Dis. 208, 1443–1447 (2013).
Nixon, C. C. et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 578, 160–165 (2020).
McBrien, J. B. et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 578, 154–159 (2020).
Borducchi, E. N. et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 540, 284–287 (2016).
Gautam, R. et al. Pathogenicity and mucosal transmissibility of the R5-tropic simian/human immunodeficiency virus SHIV(AD8) in rhesus macaques: implications for use in vaccine studies. J. Virol. 86, 8516–8526 (2012).
Okoye, A. A. et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat. Med. 24, 1430–1440 (2018).
Khanal, S. et al. In vivo validation of the viral barcoding of simian immunodeficiency virus SIVmac239 and the development of new barcoded SIV and subtype B and C simian–human immunodeficiency viruses. J. Virol. 94, e01420-19 (2019).
Elsheikh, M. M., Tang, Y., Li, D. & Jiang, G. Deep latency: a new insight into a functional HIV cure. EBioMedicine 45, 624–629 (2019).
Kessing, C. F. et al. In vivo suppression of HIV rebound by didehydro-cortistatin A, a ‘block-and-lock’ strategy for HIV-1 treatment. Cell Rep. 21, 600–611 (2017).
Margolis, D. M. et al. Curing HIV: seeking to target and clear persistent infection. Cell 181, 189–206 (2020).
Hosmane, N. N. et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J. Exp. Med. 214, 959–972 (2017).
Kok, Y. L. et al. Spontaneous reactivation of latent HIV-1 promoters is linked to the cell cycle as revealed by a genetic-insulators-containing dual-fluorescence HIV-1-based vector. Sci. Rep. 8, 10204 (2018).
Singh, A., Razooky, B., Cox, C. D., Simpson, M. L. & Weinberger, L. S. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophys. J. 98, L32–L34 (2010).
Cillo, A. R. et al. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc. Natl Acad. Sci. USA 111, 7078–7083 (2014).
Laird, G. M. et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Invest. 125, 1901–1912 (2015).
Zerbato, J. M. et al. Multiply spliced HIV RNA is a predictive measure of virus production ex vivo and in vivo following reversal of HIV latency. EBioMedicine 65, 103241 (2021).
Huang, S. H. et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J. Clin. Investig. 128, 876–889 (2018).
Gay, C. L. et al. Assessing the impact of AGS-004, a dendritic cell-based immunotherapy, and vorinostat on persistent HIV-1 Infection. Sci. Rep. 10, 5134 (2020).
Fidler, S. et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): a phase 2, randomised trial. Lancet 395, 888–898 (2020).
Gutiérrez, C. et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 30, 1385–1392 (2016).
Vibholm, L. et al. Short-course Toll-like receptor 9 agonist treatment impacts innate immunity and plasma viremia in individuals with human immunodeficiency virus infection. Clin. Infect. Dis. 64, 1686–1695 (2017).
Riddler, S. A. et al. Vesatolimod, a toll-like receptor 7 agonist, induces immune activation in virally suppressed adults with HIV-1. Clin. Infect. Dis. 72, e815–e824 (2020).
Elliott, J. H. et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV 2, 17 (2015).
McMahon, J. H. et al. Neurotoxicity with high dose disulfiram and vorinostat used for HIV latency reversal. AIDS https://doi.org/10.1097/qad.0000000000003091 (2021).
Kim, Y., Anderson, J. L. & Lewin, S. R. Getting the ‘kill’ into ‘shock and kill’: strategies to eliminate latent HIV. Cell Host Microbe 23, 14–26 (2018).
Mousseau, G. et al. The tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio 6, e00465 (2015).
Ahlenstiel, C. et al. Novel RNA duplex locks HIV-1 in a latent state via chromatin-mediated transcriptional silencing. Mol. Ther. Nucleic Acids 4, e261 (2015).
Besnard, E. et al. The mTOR complex controls HIV latency. Cell Host Microbe 20, 785–797 (2016).
Mori, L. et al. The XPB subunit of the TFIIH complex plays a critical role in HIV-1 transcription and XPB inhibition by spironolactone prevents HIV-1 reactivation from latency. J Virol. 95, e01247-20 (2020).
Timmons, A. et al. HSF1 inhibition attenuates HIV-1 latency reversal mediated by several candidate LRAs in vitro and ex vivo. Proc. Natl Acad. Sci. USA 117, 15763–15771 (2020).
Yeh, Y. J. et al. Filgotinib suppresses HIV-1-driven gene transcription by inhibiting HIV-1 splicing and T cell activation. J. Clin. Invest. 130, 4969–4984 (2020).
Gavegnano, C. et al. Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob. Agents Chemother. 58, 1977–1986 (2014).
Marconi, V. C. et al. Randomized trial of ruxolitinib in antiretroviral-treated adults with HIV. Clin. Infect. Dis. ciab212 (2021).
Henrich, T. J. et al. Everolimus, an mTORC1/2 inhibitor, in ART-suppressed individuals who received solid organ transplantation: A prospective study. Am. J. Transplant 21, 1765–1779 (2020).
Goonetilleke, N., Clutton, G., Swanstrom, R. & Joseph, S. B. Blocking formation of the stable HIV reservoir: a new perspective for HIV-1 cure. Front. Immunol. 10, 1966 (2019).
Abrahams, M. R. et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aaw5589 (2019).
Brodin, J. et al. Establishment and stability of the latent HIV-1 DNA reservoir. eLlfe 5, e18889 (2016).
Borducchi, E. N. et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 563, 360–364 (2018).
SenGupta, D. et al. The TLR7 agonist vesatolimod induced a modest delay in viral rebound in HIV controllers after cessation of antiretroviral therapy. Sci. Transl. Med. 13, eabg3071 (2021).
Mothe, B. et al. HIVconsv vaccines and romidepsin in early-treated HIV-1-infected individuals: safety, immunogenicity and effect on the viral reservoir (Study BCN02). Front. Immunol. 11, 823 (2020).
Fidler, S. et al. Antiretroviral therapy alone versus antiretroviral therapy with a kick and kill approach, on measures of the HIV reservoir in participants with recent HIV infection (the RIVER trial): a phase 2, randomised trial. Lancet 395, 888–898 (2020).
Leth, S. et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet HIV 3, e463–e472 (2016).
Conway, J. M. & Perelson, A. S. Post-treatment control of HIV infection. Proc. Natl Acad. Sci. USA 112, 5467–5472 (2015).
Hansen, S. G. et al. Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013).
Steichen, J. M. et al. A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366, eaax4380 (2019).
Rasmussen, T. et al. Impact of anti-PD-1 and anti-CTLA-4 on the HIV reservoir in people living with HIV with cancer on antiretroviral therapy: The AIDS Malignancy Consortium-095 study. Clin. Infect. Dis. 73, e1973–e1981 (2020).
Riddler, S. A. et al. Vesatolimod, a toll-like Receptor 7 agonist, induces immune activation in virally suppressed adults living with human immunodeficiency virus-1. Clin. Infect. Dis. 72, e815–e824 (2021).
Papasavvas, E. et al. Safety, immune, and antiviral effects of pegylated interferon alpha 2b administration in antiretroviral therapy-suppressed individuals: results of pilot clinical trial. AIDS Res Hum. Retroviruses 37, 433–443 (2021).
Mendoza, P. et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 561, 479–484 (2018).
Colby, D. J. et al. Safety and immunogenicity of Ad26 and MVA vaccines in acutely treated HIV and effect on viral rebound after antiretroviral therapy interruption. Nat. Med. 26, 498–501 (2020).
Salgado, M. et al. Mechanisms that contribute to a profound reduction of the hiv-1 reservoir after allogeneic stem cell transplant. Ann. Intern. Med. 169, 674–683 (2018).
Koelsch, K. K. et al. Impact of allogeneic hematopoietic stem cell transplantation on the HIV Reservoir and immune response in 3 hiv-infected individuals. J. Acquired Immune Defic. Syndromes 75, 328–337 (2017).
Yukl, S. A. et al. Challenges in detecting HIV persistence during potentially curative interventions: a study of the Berlin patient. PLoS Pathog. 9, e1003347 (2013).
Gupta, R. K. et al. Evidence for HIV-1 cure after CCR5∆32/∆32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV 7, e340–e347 (2020).
Tebas, P. et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Invest. 131, e144486 (2021).
Tebas, P. et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910 (2014).
Martinez-Navio, J. M. et al. Adeno-associated virus delivery of anti-HIV monoclonal antibodies can drive long-term virologic suppression. Immunity 50, 567–575(2019).
Priddy, F. H. et al. Adeno-associated virus vectored immunoprophylaxis to prevent HIV in healthy adults: a phase 1 randomised controlled trial. Lancet HIV 6, e230–e239 (2019).
Gardner, M. R. et al. AAV-delivered eCD4-Ig protects rhesus macaques from high-dose SIVmac239 challenges. Sci. Transl. Med. 11, eaau5409 (2019).
Mancuso, P. et al. CRISPR based editing of SIV proviral DNA in ART treated non-human primates. Nat. Commun. 11, 6065 (2020).
Karpinski, J. et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 34, 401–409 (2016).
Schuster, S. J. et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377, 2545–2554 (2017).
Herzig, E. et al. Attacking Latent HIV with convertible CAR-T Cells, a highly adaptable killing platform. Cell 179, 880–894 e810 (2019).
Rust, B. J. et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood 136, 1722–1734 (2020).
Tombacz, I. et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol. Ther. (2021).
Nahmad, A. D. et al. Engineered B cells expressing an anti-HIV antibody enable memory retention, isotype switching and clonal expansion. Nat. Commun. 11, 5851 (2020).
Huang, D. et al. Vaccine elicitation of HIV broadly neutralizing antibodies from engineered B cells. Nat. Commun. 11, 5850 (2020).
Gillmore, J. D. et al. CRISPR–Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021).
Adair, J. E. et al. Towards access for all: 1st Working Group Report for the Global Gene Therapy Initiative (GGTI). https://doi.org/10.1038/s41434-021-00284-4(2021).
Persaud, D. et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N. Engl. J. Med. 369, 1828–1835 (2013).
Luzuriaga, K. et al. Viremic relapse after HIV-1 remission in a perinatally infected child. N. Engl. J. Med. 372, 786–788 (2015).
Frange, P. et al. HIV-1 virological remission lasting more than 12 years after interruption of early antiretroviral therapy in a perinatally infected teenager enrolled in the French ANRS EPF-CO10 paediatric cohort: a case report. Lancet HIV 3, e49–e54 (2016).
Violari, A. et al. A child with perinatal HIV infection and long-term sustained virological control following antiretroviral treatment cessation. Nat. Commun. 10, 412 (2019).
Mavigner, M. et al. Simian immunodeficiency virus persistence in cellular and anatomic reservoirs in antiretroviral therapy-suppressed infant rhesus macaques. J. Virol. 92, e00562-18 (2018).
Obregon-Perko, V. et al. Simian–human immunodeficiency virus SHIV.C.CH505 persistence in ART-suppressed infant macaques is characterized by elevated SHIV RNA in the gut and a high abundance of intact SHIV DNA in naive CD4+ T cells. J. Virol. 95, e01669-20 (2020).
Bricker, K. M. et al. Therapeutic vaccination of SIV-infected, ART-treated infant rhesus macaques using Ad48/MVA in combination with TLR-7 stimulation. PLoS Pathog. 16, e1008954 (2020).
Hessell, A. J. et al. Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat. Med. 22, 362–368 (2016).
Shapiro, M. B. et al. Single-dose bNAb cocktail or abbreviated ART post-exposure regimens achieve tight SHIV control without adaptive immunity. Nat. Commun. 11, 70 (2020).
Dhummakupt, A. et al. Differences in inducibility of the latent HIV reservoir in perinatal and adult infection. JCI Insight 5, e134105 (2020).
Hill, A. L. et al. Real-time predictions of reservoir size and rebound time during antiretroviral therapy interruption trials for HIV. PLoS Pathog. 12, e1005535 (2016).
Julg, B. et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials-report of a consensus meeting. Lancet HIV 6, e259–e268 (2019).
Garner, S. A. et al. Interrupting antiretroviral treatment in HIV cure research: scientific and ethical considerations. J. Virus Erad. 3, 82–84 (2017).
Protiere, C. et al. Differences in HIV cure clinical trial preferences of French people living with HIV and physicians in the ANRS-APSEC study: a discrete choice experiment. J. Int AIDS Soc. 23, e25443 (2020).
Peluso, M. J. et al. A collaborative, multidisciplinary approach to HIV transmission risk mitigation during analytic treatment interruption. J. Virus Erad. 6, 34–37 (2020).
Dybul, M. et al. The case for an HIV cure and how to get there. Lancet HIV 8, e51–e58 (2021).
Johnston, R. E. & Heitzeg, M. M. Sex, age, race and intervention type in clinical studies of HIV cure: a systematic review. AIDS Res Hum. Retroviruses 31, 85–97 (2015).
Dube, K. et al. Considerations for increasing racial, ethnic, gender, and sexual diversity in HIV cure-related research with analytical treatment interruptions: a qualitative inquiry. https://doi.org/10.1089/AID.2021.0023 (2021).
Fidler, S. et al. HIV cure research in the time of COVID-19 — antiretroviral therapy treatment interruption trials: a discussion paper. J. Virus Erad. 7, 100025 (2021).
Acknowledgements
We acknowledge the generous contribution of all the participants in the working groups, the key opinion leaders who read and provided feedback on the strategy, participants in the online survey and secretarial support from the International AIDS Society. S.R.L. and S.G.D. are funded by National Institutes of Health Delaney AIDS Research Enterprise (DARE) Collaboratory (UM1AI126611 and UM1AI164560). S.R.L. is also funded by the National Health and Medical Research Council (NHMRC; grant number GNT1149990) of Australia and the Australian Centre for HIV and Hepatitis. R.B.J. is funded by the NIH UM1AI64565. C.T.T. is funded by the South African Research Chairs Initiative of the Department of Science and Innovation and National Research Foundation of South Africa (grant 84177). O.L. is funded by the ANRS, Sidaction, University Paris Saclay, Inserm, and CEA (Commissariat à l’Energie Atomique). P.C. is funded by the NIH (HL156247 and AI164561); N.A. is funded by the NIH Delaney CARE Collaboratory 1UM1AI126619 and from R01AI134363; T.N. is funded by the South African Research Chairs Initiative of the Department of Science and Innovation and National Research Foundation of South Africa (grant 64809), The Bill and Melinda Gates Foundation (INV-033558), the International AIDS Vaccine Initiative (UKZNRSA1001) and DFG German-African Network grant (grant number AL 1043/6-1).
Author information
Authors and Affiliations
Consortia
Contributions
S.G.D., S.R.L., M.D.J. and R.L. developed the method for generating the strategy and oversaw the governance and establishment of the working groups. All authors on the masthead were members of the steering group. All authors of the IAS Global Scientific Strategy writing group contributed to the writing and approved the submitted version of the manuscript. Members of the IAS Global Scientific Strategy working groups are identified in the list at the end of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.G.D. receives research support from Gilead and Merck. He is a member of the scientific advisory boards for BryoLogyx, Enochian Biosciences and Tendel. He has consulted for AbbVie, Biotron, Eli Lilly, GSK/ViiV and Immunocore; J.S. is a member of Merck KGaA’s Ethics Advisory Panel and Stem Cell Research Oversight Committee; a member of IQVIA’s Ethics Advisory Panel; a member of Aspen Neurosciences Clinical Advisory Panel; a member of a Merck Data Monitoring Committee; a consultant to Biogen; and a consultant to Portola Pharmaceuticals Inc. None of these activities are related to the issues discussed in this manuscript; T.N. has received research funding from Gilead Sciences; O.L. has been paid expert testimony and consultancy fees from BMS France, MSD, Astra Zeneca; consultancy fees from Incyte, Sobi, grants from ViiV and Gilead; L.V. receives research grants from J&J, ViiV Healthcare and Gilead Sciences; P.C. is a member of Gilead’s HIV Cure Advisory Board; S.R.L.’s institution receives funding for investigator initiated research from Gilead, Merck and Viiv. She has research collaborations with BMS, Abbvie and Merck. She has received honoraria paid to her for membership of advisory boards to Gilead, Merck, Viiv, Immunocore, Vaxxinity, Biotron, Esfam and Abivax; R.L. is an employee of the International AIDS Society; M.d.J. was paid as a consultant by the International AIDS Society. S.C., R.B.J., C.T. and N.A. have no interests to declare.
Additional information
Peer review information Nature Medicine thanks Ravindra Gupta and the other, anonymous, reviewers for their contribution to the peer review of this work. Karen O’Leary was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Deeks, S.G., Archin, N., Cannon, P. et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat Med 27, 2085–2098 (2021). https://doi.org/10.1038/s41591-021-01590-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01590-5
This article is cited by
-
Balancing public health and privacy rights: a mixed-methods study on disclosure obligations of people living with HIV to their partners in China
Harm Reduction Journal (2024)
-
Ripretinib inhibits HIV-1 transcription through modulation of PI3K-AKT-mTOR
Acta Pharmacologica Sinica (2024)
-
Prevention, treatment and cure of HIV infection
Nature Reviews Microbiology (2023)
-
Inhibition of the TRIM24 bromodomain reactivates latent HIV-1
Scientific Reports (2023)
-
HIV infection
Nature Reviews Disease Primers (2023)
