
Abstract
Advanced systemic mastocytosis (AdvSM) is a rare hematologic neoplasm driven by the KIT D816V mutation and associated with poor survival. This phase 1 study (NCT02561988) evaluated avapritinib (BLU-285), a selective KIT D816V inhibitor, in patients with AdvSM. The primary endpoints were the maximum tolerated dose, recommended phase 2 dose and safety of avapritinib. Secondary endpoints included overall response rate and changes in measures of mast cell burden. Avapritinib was evaluated at doses of 30–400 mg once daily in 86 patients, 69 with centrally confirmed AdvSM. Maximum tolerated dose was not reached, and 200 mg and 300 mg daily were studied in dose-expansion cohorts. The most frequent adverse events observed were periorbital edema (69%), anemia (55%), diarrhea (45%), thrombocytopenia (44%) and nausea (44%). Intracranial bleeding occurred in 13% overall, but in only 1% of patients without severe thrombocytopenia (platelets <50 × 109/l). In 53 response-evaluable patients, the overall response rate was 75%. The complete remission rate was 36%. Avapritinib elicited ≥50% reductions in marrow mast cells and serum tryptase in 92% and 99% of patients, respectively. Avapritinib induced deep and durable responses, including molecular remission of KIT D816V in patients with AdvSM, and was well tolerated at the recommended phase 2 dose of 200 mg daily.
Similar content being viewed by others
Main
Systemic mastocytosis (SM) is a rare hematologic neoplasm characterized by accumulation of neoplastic mast cells in the bone marrow (BM), skin and other organs1,2,3,4. Mast cell mediator release and SM-related organ damage (known as C-findings) lead to severe, debilitating symptoms5,6. AdvSM comprises three subtypes: aggressive SM (ASM), SM with an associated hematologic neoplasm (SM-AHN), which accounts for ~60–70% of advanced disease7,8,9,10, and mast cell leukemia (MCL)6. Median overall survival (OS) in patients with AdvSM is ≤3.5 years, usually due to organ damage and/or progression of the associated hematologic neoplasm (AHN)1,5,6,11,12,13,14.
SM is driven by the KIT p.Asp816Val (D816V) mutation in approximately 95% of cases14,15,16, yet until recently, therapies designed to specifically target KIT D816V were unavailable6,12,17. Imatinib lacks activity against KIT D816V18,19,20 and is approved in the United States for the rare patients who have ASM without KIT D816V mutation or unknown KIT mutation status21,22. The multikinase inhibitor midostaurin is approved for AdvSM; however, few patients achieve complete remission, and gastrointestinal adverse events (AEs) are common7,8,11,23,24. Safe and effective therapies for patients with AdvSM remain an area of unmet need.
Avapritinib (BLU-285, Blueprint Medicines Corporation) is a new, selective type 1 KIT inhibitor with high potency for KIT D816V12,24,25,26. The US Food and Drug Administration (FDA) approved avapritinib in June 2021 for adult patients with AdvSM, including patients with ASM, SM-AHN and MCL. It was also recently approved in the United States for the treatment of adults with unresectable or metastatic gastrointestinal stromal tumor (GIST) harboring platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutations at a recommended starting dose of 300 mg once daily (QD), based on dose-escalation and dose-expansion trials that evaluated efficacy and safety in this setting27,28. We describe the results of a phase 1 international, multicenter, open-label study (EXPLORER, NCT02561988) evaluating the safety, pharmacokinetics (PK), efficacy and patient-reported outcomes (PROs) of avapritinib in adult patients with AdvSM and other myeloid malignancies.
Results
Patients
Between 10 March 2016 and 18 March 2020, 86 patients (dose escalation, n = 32; dose expansion, n = 54) were enrolled at ten sites in the United States and the United Kingdom (Fig. 1).
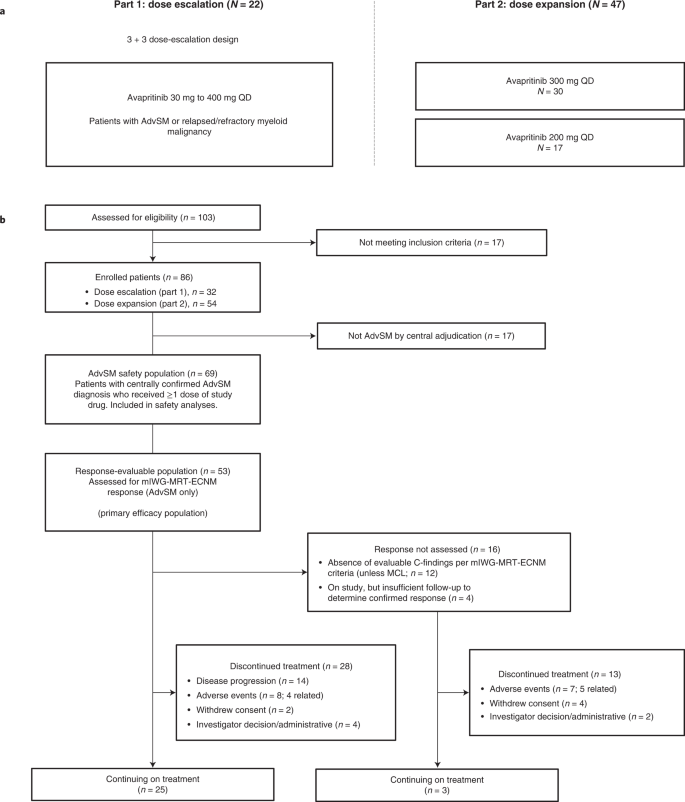
a, Study design showing patients with confirmed AdvSM. b, Patient disposition. AdvSM, advanced systemic mastocytosis; mIWG-MRT-ECNM, modified International Working Group-Myeloproliferative Neoplasms Research and Treatment and European Competence Network on Mastocytosis; MCL, mast cell leukemia; QD, once daily.
Diagnoses were centrally confirmed retrospectively by review of pathology and adjudication of C-findings using World Health Organization (WHO) criteria (Methods). Of note, previously undiagnosed AHN was identified by central pathology in 41% of patients with a local diagnosis of ASM, resulting in reclassification as SM-AHN for analysis. Overall, 69 of the 86 enrolled patients had confirmed AdvSM, while the remaining patients were adjudicated to have diagnoses of indolent SM (n = 14), smoldering SM (n = 2) or chronic myelomonocytic leukemia (n = 1). Modified International Working Group-Myeloproliferative Neoplasm Research and Treatment-European Competence Network on Mastocytosis (mIWG-MRT-ECNM) response outcomes are reported here for patients with a centrally adjudicated diagnosis of AdvSM and at least one evaluable mIWG-MRT-ECNM C-finding (the response-evaluable population), while safety is reported in all treated patients and for those with AdvSM.
At baseline, measures of mast cell disease burden (BM mast cell burden, serum tryptase level) were similar in the safety, AdvSM safety and response-evaluable populations (Table 1). The most common evaluable C-findings per mIWG-MRT-ECNM criteria at baseline were marked splenomegaly (53%), thrombocytopenia (36%), elevated alkaline phosphatase (34%), anemia (32%) and ascites (19%; Supplementary Table 1).
Forty-one of 69 patients with AdvSM (59%) had received prior antineoplastic therapy, including 23 (33%) previously treated with midostaurin and 10 (14%) with cladribine (Supplementary Table 2). Thirty-six patients with AdvSM (52%) had at least one mutation in the SRSF2, ASXL1 or RUNX1 (S/A/R) genes, which are associated with poor disease prognosis29.
Maximum tolerated dose and recommended phase 2 dose
Part 1 studied doses of 30, 60, 100, 130, 200, 300 and 400 mg QD, with doses of 200 mg and higher achieving exposures consistently above, and doses of 100 mg and 130 mg partly above, the xenograft 90% inhibitory concentration (IC90) for KIT D816V inhibition at steady state (Results and Extended Data Fig. 1). No maximum tolerated dose (MTD) was determined. During part 1, one patient experienced dose-limiting toxicity (DLT) of grade 4 vomiting at 400 mg daily. A recommended phase 2 dose (RP2D) of 300 mg QD was initially selected for use in part 2; however, dose reduction to 200 mg QD was typical, most commonly for cytopenias. Importantly, 200 mg QD had similar exposure and time to response, so a second expansion cohort with a 200 mg QD starting dose was added by protocol amendment. In total, 15 patients initiated treatment at <200 mg QD, 21 patients initiated at 200 mg QD, 43 patients initiated at 300 mg QD and 7 patients initiated at 400 mg QD.
Safety
At data cutoff, 35 of 69 patients with AdvSM (51%) had discontinued treatment: 14 (20%) due to disease progression, 7 (10%) due to related AEs, 6 (9%) due to unrelated AEs, 3 (4%) due to consent withdrawal and 5 (7%) due to investigator decision or administrative causes. The most frequent AEs are presented in Table 2, for the safety and AdvSM safety populations, and treatment-related AEs as determined by the investigator are presented in Extended Data Fig. 2.
In patients with AdvSM (n = 69), the most frequent non-hematologic AEs of any grade were periorbital edema (65%), peripheral edema (45%), diarrhea (43%), nausea (42%), fatigue (35%) and vomiting (32%). The most frequent non-hematologic AEs of grade 3 or 4 were fatigue (10%), nausea and vomiting (both 4%), and arthralgia and hypokalemia (both 3%). The incidence of non-hematologic AEs was comparable between all patients and those with confirmed AdvSM.
In patients with AdvSM, the most frequent hematologic AEs (any grade and grade 3 or 4) were anemia (58% and 36%), thrombocytopenia (51% and 41%) and neutropenia (22% and 19%). Patients with baseline cytopenias were more likely to have cytopenia AEs on study. The risk of ≥grade 3 thrombocytopenia was 20% in the absence of baseline thrombocytopenia, but 70% where thrombocytopenia was present at baseline. Neutropenia of ≥grade 3 occurred in 25% without, and 63% with baseline neutropenia. For ≥grade 3 leukopenia, the risk ratios were 67% versus 19%, with or without any baseline leukopenia, respectively.
Hair color changes, typically grade 1, occurred in 13 patients (19%), and grade 1 skin hypopigmentation was reported in two patients (3%), possibly consistent with inhibition of wild-type KIT. There was one case of anaphylactic reaction (not considered treatment related).
Cognitive effects (memory impairment, cognitive disorder, confusional state and encephalopathy) occurred in 21 patients (30%). These events were mostly grade 1 (20%) or grade 2 (7%; Extended Data Fig. 3). Grade 3 cognitive effects occurred in two patients (3%), each in the setting of concomitant benzodiazepine and/or opioid treatment. Cognitive effects were less frequent with starting doses of ≤200 mg and rarely led to treatment discontinuation (two patients; 3%).
Intracranial bleeding (ICB) was reported in nine patients with AdvSM (13%; Extended Data Fig. 3). Five of the events were asymptomatic (grade 1) and identified by prespecified imaging per protocol, two were grade 2, one was grade 3 and one was grade 5 (which was associated with head trauma). Notably, seven of the events were associated with antecedent severe thrombocytopenia (platelet count <50 × 109/l), and one additional ICB event occurred in the context of rapidly progressive MCL associated with severe thrombocytopenia. Only one (1%) patient experienced ICB in the absence of prior severe thrombocytopenia. Avapritinib dose, platelet count, coagulation studies and platelet transfusions for patients who had ICB are reported in Extended Data Fig. 4. Due to the increased incidence of ICB in association with antecedent severe thrombocytopenia, the protocol was amended to exclude the enrollment of new patients with severe thrombocytopenia (<50 × 109/l), to increase monitoring of the platelet count, and to provide guidance for pausing avapritinib and providing platelet transfusion support for severe thrombocytopenia.
Fifty (72%) patients with AdvSM experienced at least one dose reduction due to AEs (most commonly for cytopenias), with a median time to first dose reduction of 8 weeks (range, 0–109 weeks). The median daily dose was 164 mg QD (range, 30–317 mg) for all patients, which was similar for patients who started at 200 mg QD. Treatment-related AEs associated with treatment discontinuation are shown in Extended Data Fig. 5. There were six deaths due to AEs (acute myeloid leukemia, gastric hemorrhage, ICB (considered treatment related; occurring in the setting of antecedent severe thrombocytopenia and a recent fall with head trauma), cardiac arrest, staphylococcal sepsis and septic shock).
Efficacy in patients with AdvSM
Fifty-three patients with AdvSM with baseline measurable C-finding(s) were evaluable for adjudicated response assessment per mIWG-MRT-ECNM criteria; this response-evaluable population comprised 3 patients with ASM, 37 patients with SM-AHN and 13 patients with MCL. Among these patients, overall response rate (ORR) was 75% (40/53; 95% confidence interval (CI), 62–86%), with 19 (36%) exhibiting complete remission with full hematologic recovery (CR) or complete remission with partial hematologic recovery (CRh; Table 3). In addition, 18 (34%) achieved a partial remission (PR), and three (6%) demonstrated clinical improvement. The ORR was 83% (30/36; 95% CI, 67–94%) in midostaurin-naïve patients, and 59% (10/17; 95% CI, 33–82%) in patients with prior midostaurin exposure. The CR/CRh rate was 44% (16/36) in midostaurin-naïve patients and 18% (3/17) in those with prior midostaurin exposure. The CR and CRh rates were 17% (6/36) and 28% (10/36) in midostaurin-naïve patients, and 12% (2/17) and 6% (1/17) in patients with prior midostaurin exposure, respectively. The ORRs in patients with co-mutations in poor-prognosis S/A/R genes were similar to those without (74% versus 77%, respectively).
The median time to achieve PR or better (CR/CRh/PR) was 2 months (range, 2–27 months), and 9 months to achieve CR/CRh (Supplementary Fig. 1). There was a shorter median time to PR or better at starting doses of 200, 300 or 400 mg (each 2 months) compared with the median time to PR or better at starting doses of <200 mg (9 months; Extended Data Fig. 6). Median duration of response (DOR) was 38 months (95% CI, 22 months–not estimable). Estimated 12-month and 24-month DOR rates were 84% (95% CI, 72–96%) and 67% (95% CI, 49–84%), respectively.
Improvements in measures of mast cell burden in patients with AdvSM are presented in Fig. 2a–d and Extended Data Fig. 7. BM mast cells decreased by ≥50% in 92% of patients and mast cell aggregates were eliminated in 77% of patients. Serum tryptase was reduced by ≥50% in 99% of patients and reduced to <20 ng ml−1 in 74% of patients. Spleen volume was reduced by ≥35% in 82% of 66 patients with a baseline spleen volume assessment. The KIT D816V variant allele fraction (VAF) in BM was reduced from baseline by ≥50% in 80% of patients and became undetectable in 30% of patients. Clinical improvements in C-findings per mIWG-MRT-ECNM criteria observed in response-evaluable patients are presented in Supplementary Table 1.
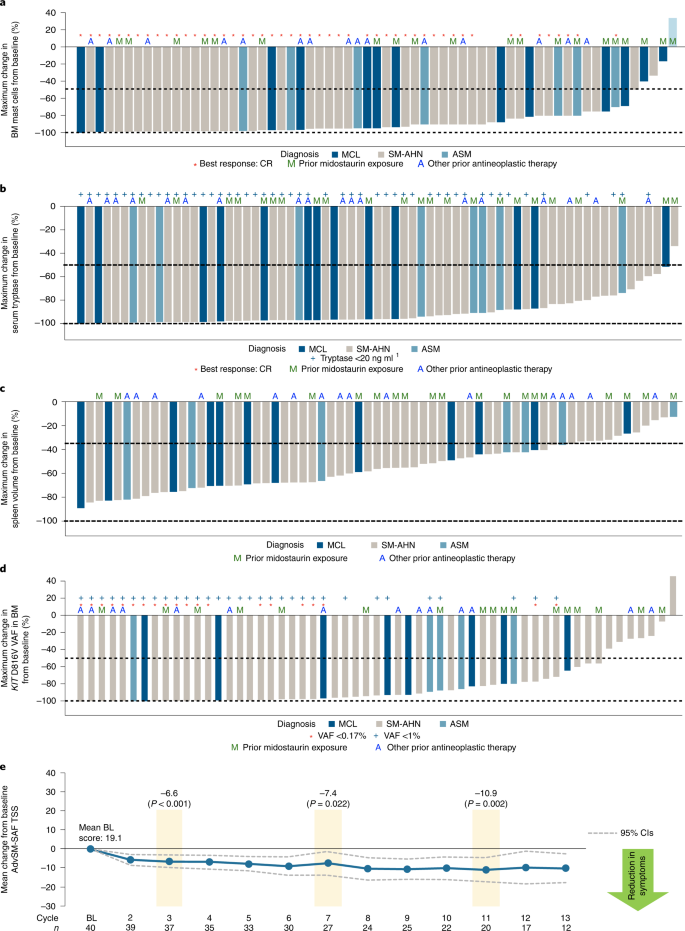
a, BM mast cell burden. b, Serum tryptase level. c, Spleen volume. d, KIT D816V VAF in BM, assessed by central ddPCR assay. e, Change from baseline in systemic mastocytosis symptom burden, evaluated by AdvSM-SAF TSS. For the change in TSS by AdvSM-SAF, a two-sided paired t-test was performed for the change from baseline at C3 (P < 0.001), C7 (P = 0.022) and C11 (P = 0.002). BL, baseline; CR, complete remission; VAF, variant allele fraction. AdvSM-SAF, advanced systemic mastocytosis symptom assessment form; ASM, aggressive systemic mastocytosis; BM, bone marrow; C, cycle; CIs, confidence intervals; ddPCR, droplet digital polymerase chain reaction; MCL, mast cell leukemia; SM-AHN, systemic mastocytosis with an associated hematologic neoplasm; TSS, total symptom score.
The depth of clinical response was generally correlated with the elimination of measurable KIT D816V allele burden in the BM, with a limit of detection of 0.17% by droplet digital polymerase chain reaction (ddPCR; Supplementary Fig. 2). A post hoc analysis according to best mIWG-MRT-ECNM response showed that complete elimination of measurable KIT D816V VAF occurred in 63% patients with a CR (n = 8), in 50% patients with a CRh (n = 8), in 23% patients with a PR (n = 13) and in only 15% of patients with a clinical improvement (n = 2) or stable disease (n = 11).
Estimated progression-free survival (PFS) rates in the response-evaluable population (n = 53) were 84% (95% CI, 73–94%) at 12 months and 63% (95% CI, 48–79%) at 24 months (Fig. 3a). During the study, six patients (9%) progressed to acute myeloid leukemia. Median OS was not reached (95% CI, 47–not estimable) in the overall AdvSM safety population (n = 69) with a median duration of follow-up of 23 months (Fig. 3b). Estimated 24-month OS rates were 76% (95% CI, 64–87%) overall, and 100%, 67% and 92% for patients with ASM, SM-AHN and MCL, respectively. OS was not substantially different for patients with or without prior midostaurin treatment (Extended Data Fig. 8); however, patients without S/A/R mutations had longer OS compared with those with S/A/R mutations (Extended Data Fig. 9). In a post hoc analysis, the mutation-adjusted risk score (MARS)30 was used for evaluation of PFS and OS risk in patients with AdvSM. MARS predicted both PFS (P = 0.0126) and OS (P = 0.0015), with a MARS ≥ 2 being associated with less favorable survival (OS shown in Supplementary Fig. 3). The OS in patients with a MARS of 0–1 was 100%.

a, PFS in the response-evaluable population. b, OS in the AdvSM safety population. AdvSM, advanced systemic mastocytosis; ASM, aggressive systemic mastocytosis; MCL, mast cell leukemia; SM-AHN, systemic mastocytosis with an associated hematologic neoplasm.
Pharmacokinetics
Steady-state mean plasma avapritinib concentration–time profiles are shown in Extended Data Fig. 1. Steady state was reached by day (D) 15. Steady-state plasma concentrations at doses of ≥200 mg QD exceeded the biochemical IC90 of 189 ng ml−1 for inhibition of KIT D816V, as measured by immunoblot in a xenograft model24,31, at most time points. Following single oral doses of 30 to 400 mg, the time to maximum plasma concentration (Tmax) ranged from 2 to 4 h. After single and repeat dosing of avapritinib, systemic exposure increased in a dose-dependent manner. The long mean plasma elimination half-life of avapritinib (range, 19.8 to 38.3 h) suggests that prolonged in vivo inhibition of KIT D816V, likely contributes to observed clinical activity, and supports QD dosing. The steady-state (cycle (C) 1, D15) geometric mean (percentage coefficient of variation; n), maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC0-τ) in patients at 200 mg QD was 433 ng ml−1 (62.2%; n = 18) and 7,340 h ng ml−1 (54.2%; n = 16), respectively.
Patient-reported outcomes and symptom improvement
The AdvSM-symptom assessment form (AdvSM-SAF) patient diary evaluated symptoms across two domains—gastrointestinal and skin. Patients provided information about the severity of abdominal pain, nausea, vomiting, diarrhea, spots, itching, flushing and fatigue, and the frequency of vomiting and diarrhea. Reductions from baseline were seen in both the gastrointestinal and skin domains. Treatment with avapritinib also yielded consistent reductions in the patient-reported AdvSM-SAF total symptom score (TSS), which encompassed gastrointestinal and skin symptoms and fatigue. The mean TSS at baseline was 19.1 points (n = 40). Statistically significant improvements in symptoms occurred rapidly and were sustained through C11 (mean change from baseline of −10.9 points, P = 0.002, n = 20; Fig. 2e).
At baseline, approximately one-third of patients overall (29 patients; 34%) were using corticosteroids. After baseline, as a result of improvement in SM-associated symptoms, 19 patients (66%) reduced their corticosteroid usage, of whom 12 (41%) patients discontinued corticosteroid use entirely, while 7 (24%) were able to reduce the dose.
Discussion
Patients with AdvSM have life-threatening organ damage and poor survival, caused by the increased proliferation of neoplastic mast cells driven by the KIT D816V mutation in ~95% of patients6. Midostaurin, until recently the only approved therapy for all subtypes of AdvSM, does not selectively target the KIT D816V mutant and only rarely do patients achieve a complete remission7. In a post hoc exploratory analysis of the pivotal midostaurin phase 2 study using IWG-MRT-ECNM criteria, responses occurred in 28% of patients with a 2% CR rate32,33.
Based on data from this phase 1 study and the ongoing phase 2 PATHFINDER (NCT03580655) study, avapritinib received United States FDA approval in June 2021 for adult patients with all subtypes of AdvSM, at a recommended dose of 200 mg orally QD. Due to the risks of ICB associated with severe thrombocytopenia, avapritinib is not recommended for the treatment of patients with AdvSM with a platelet count of <50 × 109/l. The results of this phase 1 dose-escalation/expansion study demonstrated that potent and selective inhibition of KIT D816V by avapritinib elicited rapid, profound reductions of measures of mast cell disease burden, resolved organ damage, and improved patients’ symptoms across the spectrum of AdvSM subtypes.
Responses (CR/CRh/PR/CI) occurred in 75% of patients, including 36% achieving CR/CRh. These responses were adjudicated using stringent mIWG-MRT-ECNM response criteria, which require a sustained response with 12-week confirmation and full resolution of one or more C-findings. Responses were observed irrespective of disease subtype, prior therapy or presence of high-risk S/A/R mutations.
Notably, KIT D816V became undetectable in the BM of 30% of patients with AdvSM, using a highly sensitive ddPCR assay. This demonstrated that avapritinib induced deep molecular responses, an outcome that represents a new response benchmark in AdvSM. In an earlier multivariate analysis of 38 midostaurin-treated patients with AdvSM, a mere 25% reduction in KIT D816V RNA-expressed allele burden was statistically associated with prolonged survival29.
Consistent with these findings, early outcomes with avapritinib were excellent. While midostaurin and avapritinib have not been directly compared in a prospective, randomized trial, the survival of patients with AdvSM in the current study is noteworthy. The median OS was not reached in patients with AdvSM on avapritinib, with an estimated 24-month OS rate of 76% (95% CI, 64–87%). The published literature confirms the particularly poor prognosis of patients with MCL, with only a 24% probability of 24-month OS in this subgroup34. In the current study of avapritinib, the 24-month estimated median OS in patients with MCL was 92% (95% CI, 76–100%), while in the previous trial of midostaurin, 24-month OS in patients with MCL was 26% (95% CI, 6–54%)7. Importantly, avapritinib demonstrated high ORR rates and similar OS in patients with and without prior midostaurin exposure, suggesting that cross-resistance to avapritinib is rare.
Although patients with and without S/A/R mutations at baseline had similar response rates, the presence of S/A/R mutations or a MARS score of ≥2 was associated with shorter OS. These findings suggest that although avapritinib reduced mast cell burden and C-findings regardless of co-mutation status, patients with adverse myeloid co-mutations such as S/A/R were more likely to experience disease progression. In contrast, patients with AdvSM and a MARS score of 0–1 had excellent outcomes on avapritinib monotherapy in this study, as demonstrated by a 100% 24-month OS rate.
Avapritinib at ≥200 mg QD starting doses induced rapid responses, with a median time to CR/CRh/PR of 2 months, as compared with over 9 months for <200 mg QD starting doses. Given the poor overall survival characteristic of AdvSM, the rapid responses observed with a starting dose of at least 200 mg are clinically important.
Avapritinib was generally well tolerated with few discontinuations due to AEs. The most common AEs, including fluid retention, gastrointestinal effects and cytopenias, were consistent with those reported with other inhibitors of KIT/PDGFR, and usually responded to dose modification. Cytopenias were the most common ≥grade 3 AEs, and they occurred at a higher rate in patients who had baseline cytopenias. Cognitive events and ICB have previously been reported in patients with GIST treated with avapritinib27. Although symptoms such as brain fog and memory impairment are known disease features of SM35, cognitive effect AEs were less frequent in this AdvSM population than those previously observed in patients with GIST27, which may be related to the lower starting doses in this study.
ICB was more frequent in the AdvSM setting compared with GIST and was strongly associated with antecedent severe thrombocytopenia (platelets <50 × 109/l). The incidence of ICB in AdvSM in the absence of severe thrombocytopenia (1%) was similar to the incidence in GIST. The observation of an association between severe thrombocytopenia and ICB led to the implementation of several patient selection and risk mitigation strategies in both the phase 1 EXPLORER and ongoing phase 2 PATHFINDER (NCT03580655) studies: exclusion of severely thrombocytopenic patients, increased platelet count monitoring, dose interruption in patients developing severe thrombocytopenia, and permanent treatment discontinuation after ICB of any grade. Antiplatelet agents and anticoagulants were avoided, and any coagulation abnormalities were corrected, if possible. The risk of ICB was mitigated in the aforementioned phase 2 study following the implementation of these measures. A proactive patient management strategy to minimize ICB risk will require platelet monitoring with dose interruption, reduction and platelet support in the event of severe thrombocytopenia.
The primary objectives of this dose-escalation/expansion study were to determine the MTD, safety and the RP2D of avapritinib, an investigational potent and selective KIT D816V inhibitor. Considering efficacy, tolerability and PK, the RP2D was identified as 200 mg QD based on consistent exposure above the KIT D816V in vivo IC90 and rapid time to response. At this dose, avapritinib induced deep and durable clinical, pathologic and molecular responses in patients with AdvSM, reduced disease-related symptoms, and was generally well tolerated.
The ongoing phase 2 PATHFINDER study has been designed to further characterize the safety and efficacy of avapritinib in patients with AdvSM at the 200 mg QD starting dose.
Methods
Patients
Patients aged ≥18 years with locally diagnosed AdvSM or relapsed or refractory myeloid malignancies with evidence of aberrant KIT or PDGFR signaling were enrolled in the dose-escalation phase (part 1), and only patients with locally diagnosed AdvSM according to WHO criteria36 were enrolled in the dose-expansion phase (part 2). Patients with ECOG performance status of 0–3 were eligible. Patients with platelets <25 × 109/l were excluded initially; the threshold was subsequently raised to <50 × 109/l to mitigate the risk of ICB. The complete inclusion and exclusion criteria are listed in Supplementary Table 3. Diagnoses were retrospectively adjudicated by central pathology and by an Adjudication Committee using WHO criteria. In patients with a centrally confirmed diagnosis of AdvSM, evaluable baseline C-findings and responses were defined using mIWG-MRT-ECNM13 consensus criteria (Supplementary Tables 4 and 5).
Study objectives
The primary endpoints were MTD, RP2D and assessment of safety. Secondary endpoints included the ORR based on measurable C-finding responses per mIWG-MRT-ECNM criteria, which require 12-week confirmation of response. Responses in C-findings were centrally adjudicated by a Response Adjudication Committee (RAC) comprising a subset of study investigators (see below for further details and a list of RAC members). ORR included: CR or CRh (including complete resolution of all C-findings, elimination of marrow mast cell aggregates and serum tryptase <20 ng ml−1); PR (resolution of ≥1 C-finding and ≥50% reduction in marrow mast cells and serum tryptase); and clinical improvement (resolution of ≥1 C-finding).
Other secondary endpoints included PK, DOR and changes in measures of mast cell burden (percentage of BM mast cells, serum tryptase concentration, KIT D816V VAF by ddPCR (detection limit 0.17%), and spleen and liver volumes). PROs were measured by daily completion of AdvSM-SAF (described below) for patients in part 2 as a secondary outcome37,38,39. Exploratory endpoints included time to response, PFS and OS.
Study treatment and design
This phase 1, open-label study comprised a dose-escalation phase (part 1) and a dose-expansion phase (part 2; Fig. 1). During dose escalation, patients received oral avapritinib at starting doses ranging from 30 to 400 mg QD in continuous 28-d treatment cycles. Dose escalation followed a 3 + 3 design (described below) until the MTD or RP2D was determined. During part 1, intra-patient dose escalation to assess dose levels not exceeding the MTD was allowed. Dose expansion was initially conducted at a starting dose of 300 mg QD; a second expansion cohort at 200 mg QD was subsequently introduced via protocol amendment. Dosing continued until patients experienced unacceptable toxicity, disease progression, death or withdrew consent.
3 + 3 dose-escalation study design
The 3 + 3 dose-escalation design used cohorts of three to six patients. The first cohort received avapritinib at a starting dose of 30 mg daily. Dose escalation then proceeded at increments up to 100% until one or more patients treated at a given dose level had a ≥grade 2 non-hematologic AE possibly related to avapritinib or a grade 4 hematologic AE possibly related to avapritinib, or if the given dose exceeded the highest dose determined to be safe in the first-in-human study of avapritinib in patients with GIST (400 mg). Three patients were enrolled initially in each cohort with an additional accrual of three patients if the cohort required expansion due to DLT. Once the escalation cohort was full, up to three additional patients (diagnosis of AdvSM only) could be enrolled into an enrichment cohort at a lower dose. Data from these patients were intended for further exploration of PK, pharmacodynamics and safety in patients with AdvSM.
On-study corticosteroid treatment
Corticosteroid treatment, not exceeding doses administered during screening, was permitted; doses higher than 20 mg daily prednisone equivalent were avoided, and prolonged treatment (>14 d) with high-dose corticosteroids rendered patients ineligible for response assessment.
Study oversight and review
The clinical trial registration number was NCT02561988 (date of preregistration, 15 September 2015). The study was designed by the sponsor (Blueprint Medicines) and study investigators. The full protocol was approved by the institutional review board (IRB) or independent ethics committee of each participating center: South Central–Berkshire Research Ethics Committee, Bristol; IRB, University of Pennsylvania; Dana-Farber Cancer Institute, Office for Human Research Studies; University of Utah IRB; University of Michigan Medical School IRB (IRBMED); The University of Texas MD Anderson Cancer Center IRB; Administrative Panels on Human Subjects in Medical Research, Stanford University (Stanford IRB); Western IRB (WIRB); and the Biomedical Research Alliance of New York (BRANY). The study was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonization guidelines for Good Clinical Practice and local regulations. All patients provided written informed consent. Participants were not compensated, except for reimbursement of reasonable travel expenses. All authors had access to all data, reviewed and provided critical input to the manuscript and made the decision to submit for publication. All authors vouch for the validity of the study results and adherence to the protocol.
A formal independent data monitoring committee was not used for this study. The sponsor had access to the safety data on a regular basis, as this was an open-label study. The sponsor’s clinical study team hosted investigator teleconferences on a regular basis during the study. During part 1 of the study, the clinical study team and the investigators spoke by teleconference at the end of each treatment cohort to discuss and evaluate all of the gathered safety data. At the dose-escalation teleconference, safety information, including DLTs and all grade 2 or worse AEs reported during C1, and all available PK data were described and reviewed for each patient in the current dose cohort. Updated safety, PK and other data for all other ongoing patients, including data from later cycles, were also discussed. In addition, emerging safety and efficacy data were reviewed by the sponsor at quarterly safety meetings that included the study medical monitor, site personnel and biostatisticians.
In the event a patient was withdrawn from study drug administration or the follow-up phase of the study, the medical monitor was informed. If there was a medical reason for withdrawal, the patient remained under the supervision of the investigator or designee until the condition returned to baseline or stabilized.
Progress reports and notifications of serious unexpected adverse drug reactions were provided to the IRB/independent ethics committee and regulatory authority according to local regulations and guidelines.
Study investigators
Daniel J. DeAngelo: Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA; Michael W. Deininger: Versiti Blood Research Institute and Division Hematology and Oncology, Medical College of Wisconsin, Milwaukee, WI, USA; Jason Gotlib: Stanford Cancer Institute/Stanford University School of Medicine, Stanford, CA, USA; Prithviraj Bose: The University of Texas MD Anderson Cancer Center, Houston, TX, USA; Mark W. Drummond: Beatson Cancer Centre, Glasgow, UK; Elizabeth O. Hexner: Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA, USA; Albert T. Quiery: University of Michigan, Ann Arbor, MI, USA; Deepti H. Radia: Guy’s & St Thomas’ NHS Foundation Trust, London, UK; William A. Robinson: University of Colorado, Denver, Aurora, CO, USA; Srdan Verstovsek: The University of Texas MD Anderson Cancer Center, Houston, TX, USA; Elliott F. Winton: Winship Cancer Institute, Emory University, Atlanta, GA, USA.
Central adjudication of diagnosis
To ensure a consistent diagnosis, retrospective central pathologic review of enrolled patients’ BM aspiration and biopsy samples and peripheral blood smears were assessed by an independent central pathologist. In addition, documentation (including site-entered data, as well as requested de-identified radiology reports and clinic notes) of baseline and historical (if relevant) C-findings were reviewed by the RAC to adjudicate and obtain consensus on SM diagnosis and subtyping based on the sum of available data. Diagnosis and subtyping were performed according to WHO criteria, with the following clarifications: the C-finding of weight loss was defined as documented weight loss of >10% within a 6-month (±3 months) period, and large osteolytic lesions were defined as ≥2 cm. These clarifications and strict requirement for documentation led to locally diagnosed ASM patients being reclassified as having indolent SM or smoldering SM by central diagnosis (based on data available to the RAC) for the purposes of data analysis. The MCL category also included MCL cases with evidence of AHN. The resulting comparison of local and centrally adjudicated diagnoses is presented in Supplementary Table 6.
Response adjudication committee members
Jason Gotlib (chair): Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA, USA; Daniel J. DeAngelo: Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA; Michael W. Deininger: Versiti Blood Research Institute and Division Hematology and Oncology, Medical College of Wisconsin, Milwaukee, WI, USA; Tracy I. George: ARUP Laboratories/University of Utah School of Medicine, Salt Lake City, UT, USA; Deepti H. Radia: Guy’s & St Thomas’ NHS Foundation Trust, London, UK.
Analysis populations
Eligibility was determined locally during the 56-d screening period. All patients were followed for safety or toxicity (until 30 d after treatment discontinuation) and for survival. The frequency of assessments was per protocol schedule.
The safety population included all enrolled patients and was used to report AEs and PROs (in those who completed the questionnaire). The dose-determining population included all patients in a cohort in part 1 who received ≥21 prescribed avapritinib doses in C1 and completed follow-up through C1, or experienced DLT. The determination of the MTD and RP2D were based on the dose-determining population. The AdvSM safety population comprised all patients exposed to ≥1 dose of avapritinib, who had a diagnosis of AdvSM (ASM, SM-AHN or MCL) as centrally confirmed by the RAC. The AdvSM safety population was also used to report AEs and OS. The primary efficacy (response-evaluable) population included all patients with a centrally confirmed diagnosis of AdvSM (ASM, SM-AHN or MCL) who received ≥1 dose of avapritinib, and had at least one evaluable C-finding at baseline per mIWG-MRT-ECNM criteria as adjudicated by the RAC (MCL did not require baseline C-findings), and sufficient follow-up to be assessed for confirmed response (defined as being on study for at least six 28-d treatment cycles with ≥2 post-baseline BM assessments, or having an end-of-study assessment). The response-evaluable population was used to report adjudicated response rates and PFS. The PK population comprised all patients with sufficient plasma concentration–time data to reliably estimate the PK parameters of avapritinib.
The percentage change from baseline in spleen and liver volume as measured by central radiographic assessment using an MRI or a CT scan was summarized over time in the safety population. In addition, maximum percentage reduction was presented in waterfall plots.
Spleen response was determined for patients with baseline splenomegaly (spleen ≥ 5 cm by palpation) and was assessed by palpation or spleen volume by imaging (that is, ≥35% reduction from baseline in spleen volume based on central imaging); presence of either criterion was considered a response. DOR started at the first evidence of a response in any evaluable C-finding (per mIWG-MRT-ECNM criteria) and ended at the time a response was lost (that is, loss of response per mIWG-MRT-ECNM criteria). For patients who were still responding at the time of analysis, DOR was censored at the latest time point that the patient is assessed as having clinical improvement or better. OS was analyzed in the safety population and was defined as the time from first dose to the time of death due to any cause. PK data were summarized for patients with sufficient plasma concentration–time data. Descriptive statistics were used to summarize PK parameters for each dose level, as appropriate.
No imputation was performed for missing data elements. Where the date of onset of an AE was missing, event onset was assumed to be the date of treatment to conservatively report the event as treatment emergent.
Study assessments
Responses were assessed according to the protocol schedule. Spleen size was assessed by palpation and by imaging with calculation of volume centrally. KIT D816V VAF in the BM (or in blood if unavailable) was centrally measured using a validated ddPCR assay with a lower limit of detection of 0.17% (Bio-Rad). Other somatic mutations in BM were centrally assayed at baseline by next-generation sequencing.
ORR was defined as the proportion of patients with a confirmed best response of CR, CRh, PR or clinical improvement by mIWG-MRT-ECNM consensus criteria. Time to CR/CRh/PR was defined as the time from the start of treatment to the time a CR/CRh/PR by mIWG-MRT-ECNM was first met. DOR was defined as the time from first documented response to the date of first documented progressive disease/loss of response or death due to any cause, whichever occurred first. OS was defined as the time from the start of treatment to the date of death. PFS was defined as the time from the start of treatment to the date of first documented progressive disease or death due to any cause, whichever occurred first. Progressive disease was defined per mIWG-MRT-ECNM criteria as worsening of ≥1 baseline C-finding confirmed for at least 4 weeks, or progression to acute myeloid leukemia.
Safety assessments
Safety assessments included determination of ECOG performance status, clinical laboratory testing, vital signs, electrocardiograms, brain imaging (MRI or CT scan) and physical examinations. AEs were graded according to National Cancer Institute Common Terminology Criteria (v4.03)40. Treatment-emergent AEs were defined as any AE occurring between the first dose of avapritinib through 30 d after the last dose of avapritinib, any event that was considered related to the study drug regardless of the start date of the event, or any event that was present at baseline but worsened intensity or was subsequently considered related to the study drug by the investigator.
Patient-reported outcomes assessment
The AdvSM-SAF comprises a ten-item questionnaire completed by the patient daily, specifically designed to assess symptoms related to AdvSM and measure the impact of treatments on patient symptom improvement39. It has been validated and demonstrated to be reliable and sensitive to clinical changes. The AdvSM-SAF assesses the severity of eight symptoms of AdvSM, including abdominal pain, nausea, vomiting, diarrhea, spots, itching, flushing and fatigue over a 24-h period on a numerical rating scale, and also measures the frequency of vomiting and diarrhea by asking patients to enter a discrete numerical value. Data were analyzed at the item level, domain level and total score level. Domain level analyses are:
-
Gastrointestinal symptoms score (abdominal pain, nausea, vomiting and diarrhea severity (range 0–40))
-
Skin symptom score (spots, itching and flushing severity (range 0–30))
The TSS was achieved by summing all eight severity items (range 0–80).
Pharmacokinetic assessments and analysis
On D1 and D15 of C1, blood samples for PK analysis were collected before the dose, and 0.5, 1, 2, 4, 8 and 24 h (C1D1 only) after the dose. C2D1 pre-dose concentration was used as the C1D15 24-h concentration for PK analysis. Additional pre-dose samples were collected on D1 of C3 and C4. Avapritinib PK parameters were obtained by non-compartmental analysis of the plasma concentration data versus nominal time using Phoenix WinNonlin© (version 8.0, Certara). Parameters estimated included Cmax, Tmax, AUC0–24; AUC0–τ, apparent oral clearance, apparent volume of distribution, terminal half-life, trough concentrations 24 h after dose and accumulation ratio.
Plasma concentration–time profiles across dose levels and dose versus systemic exposure (Cmax and AUC) relationships were assessed graphically. Systemic exposure parameters of interest included the arithmetic and geometric mean, Cmax and AUC, and 90% CIs.
Additional statistical methods
The total number of patients enrolled in part 1 was dependent on the observed safety profile, which determined the number of patients per dose cohort, as well as the number of dose escalations required to identify the MTD and RP2D. It was expected that approximately 25 patients who met the criteria for the dose-determining population (described below) would be enrolled in part 1. In part 2, to adequately assess the safety of avapritinib, approximately 45 patients were planned for enrollment in cohort 1 at a starting dose of 300 mg QD, and approximately 10 patients were planned to be enrolled in cohort 2 at a starting dose of 200 mg QD. With 55 patients, there is approximately 94% probability of observing an AE that occurs at a frequency of ≥5%.
Primary ORR analysis was based on RAC-adjudicated responses in the response-evaluable population. ORR was estimated using frequency, percentage and two-sided 95% CIs based on the exact binomial distribution (Clopper–Pearson). Statistical testing on binomial proportion against a null of 28% was performed using one-sided α = 0.025. Wald test P value was presented. CR + CRh + PR rate was summarized similarly as ORR. Statistical testing on binomial proportion against a null of 17% was performed using one-sided α = 0.025.
OS, PFS and DOR were determined by Kaplan–Meier estimates, and 95% CIs were estimated using Greenwood’s formula41. Comparisons between two Kaplan–Meier functions was performed using a log-rank test.
Data analysis was performed using SAS version 9.3 software.
Analysis data cutoff
The data cutoff for this analysis was 27 May 2020.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Requests for the study protocol or the anonymized derived data from this study that underlie the results reported in this article will be made available, beginning 12 months and ending 5 years following this article publication, to investigators who sign a data access agreement and provide a methodologically sound proposal to medinfo@blueprintmedicines.com.
References
Lim, K.-H. et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood 113, 5727–5736 (2009).
Hermine, O. et al. Case–control cohort study of patients’ perceptions of disability in mastocytosis. PLoS ONE 3, e2266 (2008).
Pardanani, A. et al. Prognostically relevant breakdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood 114, 3769–3772 (2009).
Akin, C. & Metcalfe, D. D. Systemic mastocytosis. Ann. Rev. Med. 55, 419–432 (2004).
Gulen, T., Hagglund, H., Dahlen, B. & Nilsson, G. Mastocytosis: the puzzling clinical spectrum and challenging diagnostic aspects of an enigmatic disease. J. Intern. Med. 279, 211–228 (2016).
Pardanani, A. Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management. Am. J. Hematol. 94, 363–377 (2019).
Gotlib, J. et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 374, 2530–2541 (2016).
DeAngelo, D. J. et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia 32, 470–478 (2018).
Reiter, A., George, T. I. & Gotlib, J. New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood 135, 1365–1376 (2020).
Schwaab, J. et al. Importance of adequate diagnostic workup for correct diagnosis of advanced systemic mastocytosis. J. Allergy Clin. Immunol. Pract. 8, 3121–3127 (2020).
Gilreath, J. A., Tchertanov, L. & Deininger, M. W. Novel approaches to treating advanced systemic mastocytosis. Clin. Pharm. 11, 77–92 (2019).
Vaes, M., Benghiat, F. S. & Hermine, O. Targeted treatment options in mastocytosis. Front. Med. (Lausanne). 4, 110 (2017).
Gotlib, J. et al. International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) & European Competence Network on Mastocytosis (ECNM) consensus response criteria in advanced systemic mastocytosis. Blood 121, 2393–2401 (2013).
Garcia-Montero, A. C. et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 108, 2366–2372 (2006).
Chatterjee, A., Ghosh, J. & Kapur, R. Mastocytosis: a mutated KIT receptor induced myeloproliferative disorder. Oncotarget 6, 18250–18264 (2015).
Orfao, A., Garcia-Montero, A. C., Sanchez, L. & Escribano, L. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br. J. Haematol. 138, 12–30 (2007).
Barete, S. et al. Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood 126, 1009–1016 (2015).
Droogendijk, H. J. et al. Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer 107, 345–351 (2006).
Pardanani, A. & Tefferi, A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood 104, 1931–1939 (2004).
Vega-Ruiz, A. et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk. Res. 33, 1481–1484 (2009).
Alvarez-Twose, I. et al. Imatinib in systemic mastocytosis: a phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget 8, 68950–68963 (2016).
US Food and Drug Administration. Gleevec (imatinib mesylate). Highlights of Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021588s024lbl.pdf (2001).
Novartis Europharm. Rydapt (midostaurin). Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product-information/rydapt-epar-product-information_en.pdf (2017).
Evans, E. K. et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 9, eaao1690 (2017).
Evans, E. et al. Blu-285, a potent and selective inhibitor for hematologic malignancies with KIT exon 17 mutations. Blood 126, 568 (2015).
Evans, E. K. et al. First selective KIT D816V inhibitor for patients with systemic mastocytosis. Blood 124, 3217 (2014).
George, S. et al. Avapritinib in patients with advanced gastrointestinal stromal tumors following at least three prior lines of therapy. Oncologist 26, e639–e649 (2021).
Heinrich, M. C. et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 21, 935–946 (2020).
Jawhar, M. et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood 130, 137–145 (2017).
Jawhar, M. et al. MARS: mutation-adjusted risk score for advanced systemic mastocytosis. J. Clin. Oncol. 37, 2846–2856 (2019).
DeAngelo, D. J., et al. Clinical activity in a phase 1 study of Blu-285, a potent, highly-selective inhibitor of KIT D816V in advanced systemic mastocytosis (AdvSM). in American Society of Hematology 59th Annual Meeting and Exposition (2017).
Tzogani, K. et al. European Medicines Agency review of midostaurin (Rydapt) for the treatment of adult patients with acute myeloid leuk/aemia and systemic mastocytosis. ESMO Open 4, e000606 (2019).
Novartis Pharmaceuticals. Rydapt (midostaurin). Full Prescribing Information. https://www.novartis.us/sites/www.novartis.us/files/rydapt.pdf (2020).
Jawhar, M. et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica 102, 1035–1043 (2017).
Jennings, S. et al. The Mastocytosis Society survey on mast cell disorders: patient experiences and perceptions. J. Allergy Clin. Immunol. Pract. 2, 70–76 (2014).
Horny H. P., et al. Mastocytosis. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Vol. 2 (eds. Swerdlow S. H. et al.) 4th edn, 586 (International Agency for Research on Cancer, 2017).
Mazar, I. et al. Development and content validity of the Advanced Systemic Mastocytosis Symptom Assessment Form (ADVSM-SAF). Value Health 19, A386 (2016).
Gotlib, J. et al. Avapritinib, a potent and selective inhibitor of KIT D816V, improves symptoms of advanced systemic mastocytosis (AdvSM): analyses of patient reported outcomes (PROs) from the phase 1 (EXPLORER) study using the (AdvSM) Symptom Assessment Form (AdvSM-SAF), a new PRO questionnaire for (AdvSM). Blood 132, 351 (2018).
Taylor, F. et al. Psychometric evaluation of the Advanced Systemic Mastocytosis Symptom Assessment Form (AdvSM-SAF). Leuk. Res. 108, 106606 (2021).
US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. Vol. 2020 (National Institutes of Health, 2009).
Klein, J. P. & Moeschberger, M. L. Survival Analysis: Techniques for Censored and Truncated Data 2nd Edn (Springer New York, 2003).
Acknowledgements
This study was funded by Blueprint Medicines Corporation. The authors thank the patients, their families and all investigators involved in this study. P.B. is supported by the National Cancer Institute (National Institutes of Health (NIH), no. P30 CA016672). J.G. is supported by an internal grant from the Stanford Cancer Institute Clinical Innovation Fund. E.O.H. is supported by the National Cancer Institute (NIH, nos. 1UE5CA246744-01 and P01-CA214278-03), the National Center for Advancing Translational Sciences (NIH no. NIH-NCATS UL1TR001878) and the National Heart, Lung and Blood Institute (NIH no. R01-HL-148014-01A1). Pathology assessment was supported by K. Karner, K. Moser and A. Rets (all ARUP Laboratories, University of Utah). Data management support was provided by C. Langford (Stanford Cancer Institute). Medical writing support was provided by Danielle Russell, PhD and Jeremy Kennard, PhD (both of Paragon, Knutsford, UK), supported by Blueprint Medicines Corporation in accordance with Good Publication Practice (GPP3) guidelines (https://www.ismpp.org/gpp3). The sponsor was involved in the study design and collection, analysis and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions and data interpretation lies with the authors.
Author information
Authors and Affiliations
Contributions
A list of study investigators and the study RAC is provided in the Methods. B.G.M. was the medical monitor of the study. H.-M.L. led the generation of data analyses. D.J.D., M.W.D. and J.G. contributed equally as senior authors. All authors were involved in data interpretation and in the writing, revision and critical review of the manuscript. All authors have approved the submitted version and are accountable for their contributions and the integrity of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: D.J.D. has served as a consultant for Amgen, Agios, Autolus, Blueprint Medicines Corporation, Forty-Seven, Incyte Corporation, Jazz, Novartis, Pfizer, Shire and Takeda, and received research funding from AbbVie, GlycoMimetics and Novartis. D.H.R. is a paid consultant for and has received research support from Blueprint Medicines Corporation, is a member of the RACs for the EXPLORER and PATHFINDER studies and has received honoraria from Novartis for educational events and consultancy. T.I.G. is a paid consultant and has served as a study steering group member for Blueprint Medicines Corporation, and is employed by ARUP Laboratories/University of Utah, which received funding from Blueprint Medicines Corporation. W.A.R. has received research funding from Blueprint Medicines Corporation. A.T.Q. has no competing interests to declare. M.W.D. received research support from Blueprint Medicines Corporation and Novartis. P.B. has received research honoraria/consulting fees from Blueprint Medicines Corporation, Incyte Corporation, Celgene Corporation (now BMS), CTI BioPharma, Kartos Therapeutics and Sierra Oncology, and research support from Blueprint Medicines Corporation, Celgene Corporation (now BMS), Kartos Therapeutics, Incyte Corporation, Constellation Pharmaceuticals, CTI BioPharma, Astellas Pharmaceuticals, Pfizer, Inc., NS Pharma and Promedior. E.O.H. has received research support and has served on a Data and Safety Monitoring Board for Blueprint Medicines Corporation, has received research support from Novartis Pharmaceuticals and Tmunity Therapeutics, and serves on the hematology exam committee for the American Board of Internal Medicine. E.F.W. has received research support for conduct of clinical studies from Blueprint Medicines Corporation, Samus Therapeutics and Incyte Corporation. H.-P.H. has served as a consultant for Novartis and Blueprint Medicines Corporation. M.T., O.S.-K. and E.K.E. are shareholders and former employees of Blueprint Medicines Corporation. H-M.L. and B.G.M. are both current employees and shareholders of Blueprint Medicines Corporation. S.V. declares research support for the conduct of clinical studies from Incyte, Roche, NS Pharma, Celgene, Gilead, Promedior, CTI BioPharma, AbbVie, Blueprint Medicines Corporation, Novartis, Sierra Oncology, PharmaEssentia, Constellation Pharmaceuticals, Ital Pharma, Protagonist and Kartos Therapeutics. M.W.D. is a paid consultant for Fusion Pharma, Takeda, Medscape, Novartis, Incyte, Sangamo, SPARC, Pfizer and DisperSol, has received research support from Blueprint Medicines Corporation and SPARC, and has served on study management committees for clinical studies sponsored by Pfizer, Takeda and Blueprint Medicines Corporation. J.G. is a paid consultant for, and has received research funding from, Blueprint Medicines Corporation, Deciphera, Incyte and Kartos Therapeutics, and has served as chair of the RAC for Blueprint Medicines Corporation’ phase 1 EXPLORER study, and for the phase 2 PATHFINDER study, and as co-chair for the Deciphera Study Steering Committee for ripretinib in AdvSM and chair of the Central Response Review Committee for the phase 2 study of bezuclastinib in AdvSM.
Additional information
Peer review information Primary Handling Editor: Saheli Sadanand, in collaboration with the Nature Medicine team. Nature Medicine thanks Jongphil Kim, Michael Mauro and Guillermo Garcia-Manero for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Steady State Mean Plasma Concentration–Time Profiles of Avapritinib in Patients with Systemic Mastocytosis.
The figure shows the mean steady state plasma concentrations of avapritinib assessed over time (Cycle 1 Day 15) in patients who received different doses of avapritinib. At most time points, avapritinib plasma concentrations at doses ≥130 mg exceeded the IC90 of 189 ng/mL for inhibition of KIT D816V measured by immunoblot in a xenograft model. Data are presented as mean values + standard deviation. The number of patients who received each dose are as follows: 30 mg, n = 3; 60 mg, n = 5; 100 mg, n = 3; 130 mg, n = 3; 200 mg, n = 21; 300 mg, n = 43; 400 mg, n = 7. h, hours; IC90, 90% inhibitory concentration; t1/2 terminal half-life.
Extended Data Fig. 2 Treatment-Related Adverse Events Reported in ≥15% of Patients.
AEs, adverse events.
Extended Data Fig. 3 Summary of Cognitive Effects and Intracranial Bleeding Events by Grade Occurring at Any Dose (AdvSM Safety Population).
*Two events were associated with head trauma and one occurred in the context of progressive mast cell leukemia. AdvSM, advanced systemic mastocytosis.
Extended Data Fig. 4 Antecedent Dose, Platelet Count, Prothrombin International Normalized Ratio (INR), Activated Partial Thromboplastin Time (aPTT) and Platelet Transfusions in Patients Who had Intracranial Bleeding Events (AdvSM Safety Population).
*Nadir platelet count presented as value during screening/nadir value within 56 days prior to event onset. †Baseline INR and aPTT presented as values during screening/maximum values within 56 days prior to event onset. ‡Total number of units of platelets given by transfusion, after first dose of avapritinib and prior to the onset of a first intracranial bleeding event. AdvSM, advanced systemic mastocytosis; aPTT, activated partial thromboplastin time; ASM, aggressive systemic mastocytosis; CTCAE, Common Terminology Criteria for Adverse Events; INR, prothrombin international normalized ratio; MCL, mast cell leukemia; NR, not recorded; RAC, Response Adjudication Committee; SM-AHN, systemic mastocytosis with an associated hematologic neoplasm.
Extended Data Fig. 5 Adverse Events Leading to Treatment Discontinuation (AdvSM Safety Population, N = 69).
*Events determined to be probably or possibly related to the study medication. †Acute myeloid leukemia is a progressive disease event that was also reported as an AE in these 3 cases. ‡Cognitive disorder and encephalopathy occurred in the same patient. §Staphylococcal sepsis and thrombocytopenia occurred in the same patient. AdvSM, advanced systemic mastocytosis.
Extended Data Fig. 6 Impact of Starting Dose on Time to Response (CR/CRh/PR) per Modified IWG-MRT-ECNM Criteria Among Responders in the Response-Evaluable Population.
CR, complete remission; CRh, CR with partial recovery of peripheral blood counts; IWG-MRT-ECNM, International Working Group-Myeloproliferative Neoplasms Research and Treatment and European Competence Network on Mastocytosis; QD, once daily; PR, partial remission.
Extended Data Fig. 7 Summary of Clinical Improvement in Measures of Mast Cell Burden (AdvSM Safety Population).
*n values indicate number of patients with neoplastic mast cell aggregates at baseline. †n values indicate number of patients with baseline serum tryptase assessment. ‡Images by CT or MRI centrally assessed by independent radiologist. §Based on central laboratory testing. AdvSM, advanced systemic mastocytosis; ASM, aggressive systemic mastocytosis; CR, complete remission; CT, computed tomography; MCL, mast cell leukemia; MRI, magnetic resonance imaging; PR, partial remission; SM-AHN, systemic mastocytosis with an associated hematologic neoplasm; VAF, variant allele fraction.
Extended Data Fig. 8 Overall Survival by Prior Midostaurin in the AdvSM Safety Population.
AdvSM, advanced systemic mastocytosis.
Extended Data Fig. 9 Overall Survival by S/A/R genotype in the AdvSM Safety Population.
a, S/A/R-positive patients. b, S/A/R-negative patients. AdvSM, advanced systemic mastocytosis; ASM, aggressive systemic mastocytosis; MCL, mast cell leukemia; SM-AHN, systemic mastocytosis with an associated hematologic neoplasm; S/A/R, SRSF2, ASXL1, or RUNX1.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3 and Supplementary Tables 1–6
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
DeAngelo, D.J., Radia, D.H., George, T.I. et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: the phase 1 EXPLORER trial. Nat Med 27, 2183–2191 (2021). https://doi.org/10.1038/s41591-021-01538-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01538-9
This article is cited by
-
Avapritinib-based SAR studies unveil a binding pocket in KIT and PDGFRA
Nature Communications (2024)
-
Systemic Mastocytosis: State of the Art
Current Hematologic Malignancy Reports (2024)
-
Cognitive Impairment and Depression in Mastocytosis: A Synthesis of the Literature
Current Allergy and Asthma Reports (2024)
-
Allogeneic Hematopoietic Cell Transplantation in Advanced Systemic Mastocytosis: A retrospective analysis of the DRST and GREM registries
Leukemia (2024)
-
Allogeneic haematopoietic cell transplantation for advanced systemic mastocytosis: Best practice recommendations on behalf of the EBMT Practice Harmonisation and Guidelines Committee
Leukemia (2024)
