
Abstract
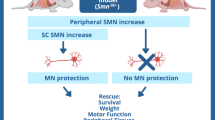
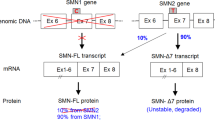
Spinal muscular atrophy type 1 (SMA1) is a debilitating neurodegenerative disease resulting from survival motor neuron 1 gene (SMN1) deletion/mutation. Onasemnogene abeparvovec (formerly AVXS-101) is a gene therapy that restores SMN production via one-time systemic administration. The present study demonstrates widespread biodistribution of vector genomes and transgenes throughout the central nervous system (CNS) and peripheral organs, after intravenous administration of an AAV9-mediated gene therapy. Two symptomatic infants with SMA1 enrolled in phase III studies received onasemnogene abeparvovec. Both patients died of respiratory complications unrelated to onasemnogene abeparvovec. One patient had improved motor function and the other died shortly after administration before appreciable clinical benefit could be observed. In both patients, onasemnogene abeparvovec DNA and messenger RNA distribution were widespread among peripheral organs and in the CNS. The greatest concentration of vector genomes was detected in the liver, with an increase over that detected in CNS tissues of 300–1,000-fold. SMN protein, which was low in an untreated SMA1 control, was clearly detectable in motor neurons, brain, skeletal muscle and multiple peripheral organs in treated patients. These data support the fact that onasemnogene abeparvovec has effective distribution, transduction and expression throughout the CNS after intravenous administration and restores SMN expression in humans.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
Data availability
Novartis is committed to sharing clinical trial data with external researchers and has been doing so voluntarily since 2014. Novartis was the third member to join ClinicalStudyDataRequest.com (CSDR), which is the first data sharing consortium of clinical study sponsors and funders. CSDR is a leader in the data sharing community inspired to drive scientific innovation and improve medical care by facilitating access to patient-level data from clinical studies. More information is available at https://www.novartisclinicaltrials.com/TrialConnectWeb/voluntarydataviewmore.nov. Due to patient consent issues, there are additional conditions for data access: (1) Participants in Novartis-sponsored trials gave their consent for the use of their data in the context of a particular trial. The external research request must therefore intend to study the medicine or disease that was intended in the original study. (2) Access to data is determined by the Independent Review Panel based on the scientific merit of the research proposal. In exceptional circumstances, access to data may be declined by the sponsor, for example, when there is a potential conflict of interest or an actual or potential competitive risk. The Independent Review Panel is coordinated by the Welcome Trust.
References
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 13, 155–165 (1995).
Burghes, A. H. M. & McGovern, V. L. in Molecular and Cellular Therapies for Motor Neuron Diseases (eds. Boulis, N. M. et al.) 121–139 (Elsevier, 2017).
Kolb, S. J. & Kissel, J. T. Spinal muscular atrophy. Neurol. Clin. 33, 831–846 (2015).
Monani, U. R. et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 (1999).
Lorson, C. L., Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA 96, 6307–6311 (1999).
Cartegni, L. & Krainer, A. R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 30, 377–384 (2002).
Lorson, C. L. et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 19, 63–66 (1998).
Burnett, B. G. et al. Regulation of SMN protein stability. Mol. Cell. Biol. 29, 1107–1115 (2009).
Lefebvre, S. et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269 (1997).
Coovert, D. D. et al. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6, 1205–1214 (1997).
Feldkötter, M., Schwarzer, V., Wirth, R., Wienker, T. F. & Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70, 358–368 (2002).
Mailman, M. D. et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 4, 20–26 (2002).
Roberts, D. F., Chavez, J. & Court, S. D. The genetic component in child mortality. Arch. Dis. Child. 45, 33–38 (1970).
D’Amico, A., Mercuri, E., Tiziano, F. D. & Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 6, 71 (2011).
Finkel, R. S. et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 83, 810–817 (2014).
Finkel, R. S. Electrophysiological and motor function scale association in a pre-symptomatic infant with spinal muscular atrophy type I. Neuromuscul. Disord. 23, 112–115 (2013).
Crawford, T. O. & Pardo, C. A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97–110 (1996).
Swoboda, K. J. et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol. 57, 704–712 (2005).
Lutz, C. M. et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 121, 3029–3041 (2011).
Le, T. T. et al. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 20, 3578–3591 (2011).
Duque, S. I. et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann. Neurol. 77, 399–414 (2015).
Farrar, M. A. et al. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 81, 355–368 (2017).
Wood, M. J. A., Bowerman, M. & Talbot, K. Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet. 26, R151–R159 (2017).
Foust, K. D. et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65 (2009).
Duque, S. et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 17, 1187–1196 (2009).
Lykken, E. A., Shyng, C., Edwards, R. J., Rozenberg, A. & Gray, S. J. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J. Neurodev. Disord. 10, 16 (2018).
Hudry, E. & Vandenberghe, L. H. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron 101, 839–862 (2019).
Bevan, A. K. et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 19, 1971–1980 (2011).
Xu, L. et al. CMV-beta-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1 alpha promoter and results in therapeutic levels of human factor X in mice. Hum. Gene Ther. 12, 563–573 (2001).
Wang, Z. et al. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene. Ther. 10, 2105–2111 (2003).
Hinderer, C. et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 29, 285–298 (2018).
Van Alstyne, M. et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. https://doi.org/10.1038/s41593-021-00827-3 (2021).
Day, J. W. et al. Clinical trial and postmarketing safety of onasemnogene abeparvovec therapy. Drug Saf. https://doi.org/10.1007/s40264-021-01107-6 (2021).
Al-Zaidy, S. A. et al. AVXS-101 (onasemnogene abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J. Neuromuscul. Dis. 6, 307–317 (2019).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Mendell, J., et al. Five-year extension results of the Phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 78, 834–841 (2021).
Day, J. W. et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy with two copies of SMN2 (STR1VE): an open-label, single-arm, phase 3 study. Lancet Neurol. 20, 284–293 (2021).
Strauss, K. et al. Onasemnogene abeparvovec gene therapy in presymptomatic spinal muscular atrophy (SMA): SPR1NT study update in children with 2 copies of SMN2. Neurology 96, S15 (2021).
Mercuri, E. et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 20, 838–841 (2021).
Zincarelli, C., Soltys, S., Rengo, G. & Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16, 1073–1080 (2008).
Blatnik, A. J. III et al. Conditional deletion of SMN in cell culture identifies functional SMN alleles. Hum. Mol. Genet. 29, 3477–3492 (2020).
Sheng, L., et al. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 48, 2853–2865 (2020).
Buchlis, G. et al. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 119, 3038–3041 (2012).
Bartus, R. T. et al. Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with alpha-synucleinopathies. Neurobiol. Dis. 78, 162–171 (2015).
Castle, M. J. et al. Postmortem analysis in a clinical trial of AAV2-NGF gene therapy for Alzheimer’s disease identifies a need for improved vector delivery. Hum. Gene. Ther. 31, 415–422 (2020).
Meyer, K. et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol. Ther. 23, 477–487 (2015).
Kolb, S. J. et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 82, 883–891 (2017).
Rindt, H. et al. Astrocytes influence the severity of spinal muscular atrophy. Hum. Mol. Genet. 15, 4094–4102 (2015).
Vukojicic, A. et al. The classical complement pathway mediates microglia-dependent remodeling of spinal motor circuits during development and in SMA. Cell. Rep. 3, 3087–3100 (2019).
Bevan, A. K. et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 19, 3895–3905 (2010).
Shababi, M. et al. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 19, 4059–4071 (2010).
Heier, C. R., Satta, R., Lutz, C. & DiDonato, C. J. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet. 19, 3906–3918 (2010).
Iascone, D. M., Henderson, C. E. & Lee, J. C. Spinal muscular atrophy: from tissue specificity to therapeutic strategies. F1000Prime Rep. 7, 4 (2015).
Wijngaarde, C. A. et al. Cardiac pathology in spinal muscular atrophy: a systematic review. Orphanet. J. Rare Dis. 12, 67 (2017).
Prior, T. W. et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 85, 408–413 (2009).
Bernal, S. et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 47, 640–642 (2010).
Oskoui, M., Darras, B.T., and De Vivo, D.C. in Spinal Muscular Atrophy (eds. Sumner, C. J. et al.) 3–19 (Academic Press, 2017).
Chand, D. et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J. Hepatol. 74, 560–566 (2021).
Feldman, A. G. et al. Subacute liver failure following gene replacement therapy for spinal muscular atrophy type I. J. Pediatr. 225, 252–258 (2020).
Acknowledgements
The authors thank and are indebted to the families and patients for making this research possible. Writing and editorial support for the original manuscript was provided by Cadent Medical Communications (New York, NY, USA). Funding was provided by Novartis Gene Therapies, Inc.
Author information
Authors and Affiliations
Contributions
G.T., A.H.M.B., C.H., J.D., B.T.T.C., S.P., B.B., W.K.C., V.L.M., R.F.H., M.C. and C.R.P. performed the experiments. G.T., A.H.M.B., P.K., D.M.S., D.E.F., W.K.C., V.L.M., M.S., F.M., J.R.M. and K.D.F. analyzed the results. K.D.F. supervised the studies.
Corresponding author
Ethics declarations
Competing interests
J.D., S.P. and B.T.T.C. are employees of Novartis Gene Therapies, Inc. and hold shares in Novartis. B.B., G.T., C.H., D.M.S., D.E.F., P.K. and K.D.F. are former employees of Novartis Gene Therapies, Inc. W.K.C. received honoraria for participation in a scientific advisory board for Regeneron Genetics Center and received grant support from Biogen for a newborn screening study outside of this study. A.H.M.B. is a consultant for Novartis Gene Therapies, Inc., for this study and other work, and conducted research for Exicure, Inc., outside of this study. V.L.M., C.R.P., and M.C. have nothing to disclose. R.F.H. was a consultant for Cunningham Meyer & Vedrine, PC, and Phillips Parker Orbeson & Arnett, PLC, outside of this study. J.R.M. is a consultant for Novartis Gene Therapies, Inc., for this study and other work, and receives honoraria for participation in scientific advisory boards from Novartis Gene Therapies, Inc., for this study and other work. M.S. reports honoraria for participation in scientific advisory boards from Novartis Gene Therapies, Inc., for this study and other work, Biogen and Roche, and for scientific presentation in Biogen-sponsored symposia outside of this study. F.M. reports a grant from Biogen for newborn screening and for registries outside of this study. F.M. also reports honoraria for participation in scientific advisory boards from Novartis Gene Therapies, Inc., for this study and other work, Biogen and Roche, and for scientific presentation at their industry-sponsored symposia outside of this study. Novartis Gene Therapies, Inc., sponsored the STR1VE-US (NCT03306277) and STR1VE-EU studies (NCT03461289).
Additional information
Peer review information Nature Medicine thanks Paola Leone and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Jerome Staal was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Tables 1–3, Figs. 1–4 and reference.
Rights and permissions
About this article

Cite this article
Thomsen, G., Burghes, A.H.M., Hsieh, C. et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat Med 27, 1701–1711 (2021). https://doi.org/10.1038/s41591-021-01483-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01483-7
This article is cited by
-
Promoting expression in gene therapy: more is not always better
EMBO Molecular Medicine (2024)
-
Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy
Nature Biomedical Engineering (2023)
-
Curing SMA: Are we there yet?
Gene Therapy (2023)
-
Safety Concerns with Nusinersen, Risdiplam, and Onasemnogene Abeparvovec in Spinal Muscular Atrophy: A Real-World Pharmacovigilance Study
Clinical Drug Investigation (2023)
-
Early treatment is a lifeline for infants with SMA
Nature Medicine (2022)
