
Abstract
More than 190 vaccines are currently in development to prevent infection by the novel severe acute respiratory syndrome coronavirus 2. Animal studies suggest that while neutralizing antibodies against the viral spike protein may correlate with protection, additional antibody functions may also be important in preventing infection. Previously, we reported early immunogenicity and safety outcomes of a viral vector coronavirus vaccine, ChAdOx1 nCoV-19 (AZD1222), in a single-blinded phase 1/2 randomized controlled trial of healthy adults aged 18–55 years (NCT04324606). Now we describe safety and exploratory humoral and cellular immunogenicity of the vaccine, from subgroups of volunteers in that trial, who were subsequently allocated to receive a homologous full-dose (SD/SD D56; n = 20) or half-dose (SD/LD D56; n = 32) ChAdOx1 booster vaccine 56 d following prime vaccination. Previously reported immunogenicity data from the open-label 28-d interval prime-boost group (SD/SD D28; n = 10) are also presented to facilitate comparison. Additionally, we describe volunteers boosted with the comparator vaccine (MenACWY; n = 10). In this interim report, we demonstrate that a booster dose of ChAdOx1 nCoV-19 is safe and better tolerated than priming doses. Using a systems serology approach we also demonstrate that anti-spike neutralizing antibody titers, as well as Fc-mediated functional antibody responses, including antibody-dependent neutrophil/monocyte phagocytosis, complement activation and natural killer cell activation, are substantially enhanced by a booster dose of vaccine. A booster dose of vaccine induced stronger antibody responses than a dose-sparing half-dose boost, although the magnitude of T cell responses did not increase with either boost dose. These data support the two-dose vaccine regime that is now being evaluated in phase 3 clinical trials.
Similar content being viewed by others


Main
Of 198 coronavirus disease 2019 (COVID-19) vaccine candidates at various developmental stages, 44 are in clinical trials, 10 of which are in late-stage clinical development1. The majority of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccine candidates in development are designed to induce immune responses against the spike surface antigen with many demonstrating early encouraging immunogenicity readouts from clinical trials2,3,4,5,6.
Much work has been carried out to characterize the immune response to infection with SARS-CoV-2 and in this manner delineate potential correlates of protection. Importantly, in a rhesus macaque challenge model, neutralizing antibody (NAb) levels following vaccination using the spike antigen correlated with protection against SARS-CoV-2 (refs. 7,8). It is therefore generally accepted that NAb levels against spike protein are likely to be critically important in protecting against overt disease, and these antibodies are detected in many but not all convalescent individuals. The magnitude of antibody responses appears to relate to antigen load, with higher responses associated with more severe disease, especially in older adults9,10,11,12. Beyond direct neutralization through blocking of viral host cell entry, other antibody functions against SARS-CoV-2 are likely to be important determinants of the course of infection. Systems serology approaches have demonstrated differences in the induction of anti-spike (SARS-CoV-2 spike protein) antibody subclasses and Fc-mediated functions between protected and susceptible animals following vaccination7,13, as well as between convalescent and deceased patients when sampled early in disease14. Anti-spike antibodies with diverse effector functions including antibody-dependent neutrophil phagocytosis, antibody-dependent natural killer (NK) cell degranulation, antibody-dependent complement deposition and antibody-dependent cellular phagocytosis were induced following initial SARS-CoV-2 infection in rhesus macaques that were subsequently protected against reinfection13. Similarly, anti-spike antibodies with broad functionalities were enriched in convalescent compared with deceased individuals14. Robust T cell responses against SARS-CoV-2 have been described during acute disease and in recovery15. Other studies have also demonstrated a highly activated cytotoxic T cell phenotype in acute disease and a polyfunctional response during convalescence16. It should be noted, however, that vaccine-mediated immune correlates of protection have yet to be defined for SARS-CoV-2 or any human coronaviruses.
Although clinical trials of multiple SARS-CoV-2 vaccine candidates2,3,4,6,12,17,18,19 have reported the induction of antibodies that bind to the spike protein, including NAbs, at the time of writing, no detailed characterization of vaccine-induced antibodies has thus far been reported in humans. We recently reported preliminary safety and immunogenicity of the adenoviral vector-based SARS-CoV-2 vaccine, ChAdOx1 nCoV-19 (AZD1222)17. Here we describe further evaluation of the quantity and quality of humoral and cellular immune responses after vaccination of individuals recruited to the phase 1/2 clinical trial of the ChAdOx1 nCoV-19 vaccine. We compare immunogenicity and reactogenicity in trial participants with different booster doses and schedules.
Results
Participants
Following a trial amendment on 22 June 2020, 52 participants aged between 18 and 55 years, who had already received an initial standard dose of ChAdOx1 nCoV-19, were boosted with a second dose of ChAdOx1 nCoV-19 at a 56-d interval. Twenty of these participants received a standard dose boost (SD/SD D56) and 32 received a dose-sparing half-dose (low-dose) boost (SD/LD D56). To maintain blinding, ten volunteers in the comparator vaccine arm also received a booster dose of the meningococcal ACWY (MenACWY) vaccine at day 56 (Supplementary Fig. 1). We previously reported on adverse events, anti-spike binding and NAb titers and interferon (IFN)-γ ELISpot responses in ten participants who received two standard doses 28 d apart (SD/SD D28)17. These data are reproduced below to enable comparison between day-28 and day-56 boost intervals. Recruitment for the trial took place between 23 April and 21 May 2020, and no withdrawals have occurred from these groups to date. Analysis of baseline blood samples from the entire trial population (n = 1,077) showed that thirty ChAdOx1 nCoV-19 prime recipients were seropositive to SARS-CoV-2 spike protein (n = 30/544) before vaccination.
Safety and tolerability
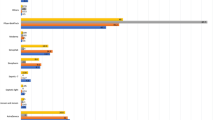
As previously reported, systemic reactogenicity following ChAdOx1 nCoV-19 prime vaccination was observed17 with 15 of 52 (28.8%) participants reporting pyrexia after vaccination (Fig. 1). However, a booster dose of ChAdOx1 nCoV-19 in the same participants resulted in minimal reactogenicity, with no episodes of pyrexia occurring (0/52). Additionally, the rates of volunteers experiencing any moderate or severe solicited systemic reactions within 7 d of vaccination were higher following prime (moderate: 27/52, 51.9%; severe; 10/52, 19.2%) compared with boost (moderate: 10/52, 19.2%; severe: 2/52, 3.8%) vaccinations. Local reactions were also higher following prime vaccinations, with moderate local reactions reported in 12 of 52 (23.1%) participants following prime compared with only 2 of 52 (3.8%) following boost vaccinations. These results are consistent with the previously reported open-label 28-d interval ChAdOx1 boost group (SD/SD D28; n = 10)17.

Local (top) and systemic (bottom) solicited reactions following prime and boost vaccinations at different dose regimens. Day 0, day of vaccination; P, post-vaccination observation period in the clinic. Fever was classified as mild (38.0 °C to <38.5 °C), moderate (38.5 °C to <39.0 °C) and severe (≥39.0 °C). Feverish was defined as a self-reported feeling of feverishness.
No serious adverse events have occurred in either SD/SD D56, SD/LD D56 or SD/SD D28 ChAdOx1 nCoV-19 prime-boost groups to date. The overall reactogenicity profile after the second dose was lower than after the first, and the pattern was consistent between groups with different doses and time intervals between doses. The most frequently reported local and systematic reactions were tenderness and fatigue, respectively (Fig. 1). We also report here local and systemic reactions for participants that received ChAdOx1 nCoV-19 and were found to be seropositive to SARS-CoV-2 spike antigen at baseline on a Meso Scale Discovery (MSD) multiplexed immunoassay (MIA) platform (n = 30; Extended Data Fig. 1). There was no appreciable difference among those who were seropositive compared with those who were seronegative at baseline in either local or systemic reactogenicity (Extended Data Fig. 2).
Meso Scale discovery multiplexed assay and neutralizing antibody titers
Anti-spike IgG antibodies to SARS-CoV-2 spike and receptor-binding domain (RBD) were measured in a serology MIA. In both cases, antibody titers rose after the first vaccination, with a further increase after the second (Fig. 2a). At 14 d after the second dose, anti-spike IgG titers were not significantly different between those who received the booster at 28 d (geometric mean titer (GMT): 35,990; 95% confidence interval (CI): 24,408–53,068; previously published as the median and interquartile range (IQR) in Folegatti et al.17) or at 56 d (SD/SD D56, GMT: 44,485, 95% CI: 31,714–62,400, P = 0.426; SD/LD D56, GMT: 25,667, 95% CI: 18,814–35,015, P = 0.250). However, those who received a half-dose boost had lower titers 14 d after the boost than those who received the standard dose (P = 0.020). Similar findings were also seen for anti-RBD IgG using MIA (Fig. 2a). Following prime vaccination, anti-spike IgG titers were increased tenfold in these individuals after 28 d (Fig. 2a and Supplementary Table 2).

a, Multiplex SARS-CoV-2 IgG response by MIA after prime-boost vaccination. Time course of IgG responses are shown for three ChAdOx1 nCoV-19 prime-boost groups; SD/SD: two standard doses administered either 28 (n = 10 participants, except for on day 42 where samples from only 9 participants were available) or 56 (n = 20 participants, except for on days 28, 70 and 84 where samples from only 19 participants were available) days apart, SD/LD: standard dose prime followed by low-dose boost 56 d apart (n = 32 participants, except for on days 70 and 84 where samples from only 29 and 31 participants were available, respectively) and for two doses of MenACWY comparator vaccine (n = 10 participants). Dashed vertical lines show time points at which boosting occurred. Plot shows the median and IQR. AU ml−1, arbitrary units per milliliter. Left: anti-RBD responses. Right: anti-spike protein responses. The dashed line indicates responses in 30 participants who received only one dose of ChAdOx1 nCoV-19 and were seropositive at baseline (seropositivity threshold defined as anti-spike IgG > 1,000 AU ml−1). b, Live SARS-CoV-2 MNA80 after prime-boost vaccination. Time course of microneutralization titer at IC80 is shown for three ChAdOx1 nCoV-19 prime-boost groups; SD/SD: two standard doses administered either 28 (n = 10 participants, except for day 42 time point where samples from only 9 participants were available) or 56 (n = 19 participants) days apart; SD/LD: standard dose prime followed by low-dose boost 56 d apart (n = 24 participants) and for two doses of MenACWY comparator (n = 10 participants). Error bars show medians and IQRs. To normalize data across assay runs, a reference sample was included in all assay runs and test samples normalized to this value by generating log10 ratios. Dashed lines show the upper limit of assay (values outside this range set to 640). The lower limit of the normalized data was set to 5. c, IFN-γ ELISpot response to peptides spanning the SARS-CoV-2 spike vaccine insert after vaccination with ChAdOx1 nCoV-19: the total ex vivo T cell response to the SARS-CoV-2 spike vaccine insert encoded within the vaccine is shown over time (IFN-γ ELISpot; spot-forming cells (SFCs) per 106 peripheral blood mononuclear cells (PBMCs), calculated by summing the responses to peptide pools corrected for background; Methods). Response is shown as the median with IQR per vaccination regimen; D56 (n = 4 at days 0 and 28, n = 10 participants at day 35, n = 20 at day 70 and n = 19 at day 84); SD/LD: standard dose prime and low-dose boost vaccination (n = 9 for days 0 and 28, n = 31 at day 70 and n = 32 at day 84). Participants received a full or half-dose booster dose of ChAdOx1 nCoV-19 at day 56 (SD/SD D56, and SD/LD D56). The lower limit of detection is 48 SFCs per 106 PBMCs and is denoted by a dashed line. MenACWY (n = 79 at day 0, n = 43 at day 7, n = 44 at day 14, n = 69 at day 28, n = 42 at day 56 and n = 10 at days 70 and 84). d, NAb levels measured in pseudovirus assay (Monogram IC50) pseudovirus neutralization titers. Dots represent individual data points. Box plots represent median titers and IQR. Left: standard dose given 28 d apart (n = 10). Middle: standard dose given 56 d apart (n = 20). Right: dose-sparing regime (standard followed by half dose) given 56 d apart (n = 32). SD/SD D28: D0, n = 10; D28, n = 10; D35, n = 7; D42, n = 9. SD/SD D56: D0, n = 20; D28, D35 and D42 not performed; D56, n = 20; D70, n = 19. SD/LD D56: D0, n = 32; D28, D35 and D42 not performed; D56, n = 32; and D70, n = 29. Data for SD/SD D28 (a–d) were previously published and reproduced here from Folegatti et al.17 with permission from Elsevier.
NAbs were assayed using a microneutralization assay (MNA) reporting the reciprocal of the serum dilution required to reduce live SARS-CoV-2 infection of single cells by 80% (MNA80). NAbs were induced following prime vaccinations and significantly increased after a booster dose in ChAdOx1 nCoV-19 groups. Median normalized NAb titers at 14 d after the boost were 274 (IQR: 232–542) for SD/SD D28, 170 (IQR: 226–368) for SD/LD D56 and 395 (IQR: 259–640) for SD/SD D56 (Fig. 2b and Supplementary Table 3). No NAb activity was observed in the MenACWY group. NAbs were also determined in a pseudovirus neutralization assay reporting the half-maximum inhibitory concentration (IC50). Median NAb titers measured with the pseudovirus assay at 14 d after boost vaccination were 253 (IQR: 100–391) for SD/LD D56 and 424 (IQR: 229–915) for SD/SD D56 (previously reported median (IQR) for for SD/SD D28: 451 (212–627))17 (Fig. 2d and Supplementary Table 4).
Anti-spike antibody class and subclass
Antibody classes and subclasses within the anti-spike response were determined. Vaccination with ChAdOx1 nCoV-19 increased anti-spike IgM and IgA titers with a peak response measured 28 d after prime for IgM. There was no difference in the response measured 14 d after SD or LD boost, and the response after boost vaccination was of similar magnitude to SD/SD with a 28-d interval (Fig. 3a). Serum samples that were IgG positive 28 d after prime vaccination were assayed for anti-spike IgG subclasses. IgG1 and IgG3 responses were readily detectable at day 28 and were at a similar level, before boosting, on day 56 in regimens with a 56-d interval. Following booster vaccination, the median IgG1 response did not increase in participants who received the standard dose regimen with a 28-d interval, although this may have been limited by the small group size (n = 10). IgG1 responses did increase 14 d after a SD or LD boost in regimens with a 56-d interval, with no measured difference in the magnitude of the response due to dose. IgG3 responses were increased following booster vaccination across all three regimens regardless of interval or dose. The response was predominantly IgG1 and IgG3, with low levels of IgG2 and IgG4 (Fig. 3b). This type-1 helper T cell-biased IgG response is in agreement with other studies investigating adenoviral vector-based vaccine priming in humans20,21,22. These analyses highlight the similarity in antibody response induced after ChAdOx1 nCoV-19 vaccination regardless of interval or booster dose20. Note that antibody isotype data for SD/SD D28 (n = 10) is also presented in Ewer et al.23 to facilitate comparison with other groups presented in that paper.

a, SARS-CoV-2 spike-specific immunoglobulin isotype responses induced by prime-boost regimens of ChAdOx1 nCoV-19. b, SARS-CoV-2 spike-specific IgG subclass responses induced by prime-boost regimens of ChAdOx1 nCoV-19. Volunteers received a SD of ChAdOx1 nCoV-19 at day 0 followed by a second vaccination with SD at day 56 (left) or LD at day 56 (middle) or SD at day 28 (right) of ChAdOx1 nCoV-19. b, SARS-CoV-2 spike trimer-specific IgA and IgM responses were quantified by ELISA and expressed as ELISA units (EUs). Solid lines connect samples from the same participant. Bold solid lines show the median with IQR. Different groups had different planned time points for blood sampling. Assays were performed at D0, D28, D56 and D70 (SD/SD D56: n = 20 participants, except for D28 where samples from only 19 volunteers were available; and SD/LD D56: n = 32 participants, except for D70 where samples from only 31 volunteers were available) or D0, D7, D14, D28, D35, D42 or D56 (SD/SD D28: n = 10 participants except for D42 where samples from only n = 9 participants were available). b, Volunteers with measurable SARS-CoV-2 spike-specific IgG at day 28 were assayed for IgG subclasses. Assays were performed at D28, D56 and D70 (SD/SD D56: n = 20 participants, except for D28 where samples from only 19 volunteers were available; and SD/LD D56: n = 32 participants, except for D70 where samples from only 31 volunteers were available) or D7, D14, D28, D35, D42 or D56 (SD/SD D28: n = 10 participants except for D42 where samples from n = 9 participants were available). SARS-CoV-2 spike-specific antibody responses were quantified by ELISA. IgG1 and IgG3 responses were expressed as EUs and IgG2 and IgG4 responses expressed as optical density (OD) at 405 nm. Solid lines connect samples from the same participant. Bold solid lines show the median with IQR.
Anti-spike antibody functionality
Anti-spike antibody function was explored further to determine the ability of antibodies induced by vaccination to support antibody-dependent monocyte phagocytosis (ADMP) and antibody-dependent neutrophil phagocytosis (ADNP). Both functions were induced by the first vaccination and substantially increased by the second, with a trend towards a larger increase when the interval between the doses was 56 d rather than 28 d, and when the booster dose was SD rather than LD (Fig. 4a,b). In comparison with serum and plasma samples taken from convalescent patients with COVID-19 between 28 and 91 d after a positive PCR test, both ADMP and ADNP were higher in the vaccinated group after the second dose. Biobanked serum samples collected from healthy volunteers before 2020 were negative in both assays, and there was no change in these functions in participants who received the MenACWY vaccine (Supplementary Table 5).

a–d, ADMP (a), ADNP (b), complement deposition (c) and NK cell activation (d) in trial participants, convalescent plasma and plasma samples donated before the pandemic began. Anti-spike antibody attributes at 14 d after boost vaccination in trial participants (e–g) and convalescent plasma (h). Longitudinal Fc-dependent antibody functionality in ChAdOx1 nCoV-19 vaccine recipients, in convalescent patients with COVID-19 and in samples collected before the pandemic. a, ADMP scores for vaccine recipients who received either two standard doses 28 d apart (SD/SD D28; n = 10) or 56 d apart (SD/SD D56; n = 19), or one standard and one low dose 56 d apart (SD/LD D56; n = 24). Convalescent patients with COVID-19 (conv; n = 48) and pre-pandemic controls (pre-2020; n = 19) are also shown. The median and IQR of normalized responses are shown for each time point studied. b, ADNP scores for vaccine recipients, convalescent patients with COVID-19 and pre-pandemic controls (SD/SD D28, n = 10; SD/SD D56, n = 18; SD/LD D56, n = 24; conv, n = 45; pre-2020, n = 14). The median and IQR of normalized responses are shown for each time point studied. c, ADCD. Background was subtracted from the MFI median and IQR for vaccine recipients who received two standard doses 56 d apart (n = 10) or one standard and one low dose 56 d apart (n = 12). Convalescent patients with COVID-19 (Conv, n = 37) are also shown. d, ADNKA. The median and IQR of the percentage of CD107a+ NK cells compared to control wells is shown for vaccine recipients who received either two standard doses 28 d apart (n = 10) or 56 d apart (n = 19), or one standard and one low dose 56 d apart (n = 22), and convalescent patients with COVID-19 (n = 28) and pre-2020 controls (n = 16). e–h, Polar plots of data normalized across all time points and groups using min–max normalization. Each wedge represents an anti-spike antibody isotype/subtype or function. For boosted groups (e–g) the median value of each antibody assay at 14 d after the booster vaccine is displayed and is represented by the size of the wedge. Note that ADCD was not performed on the SD/SD D28 group and was omitted from c and e. Values obtained from convalescent samples taken from a group of patients and asymptomatic individuals between 28 and 91 d from those with PCR-positive results are also shown (h).
Antibody-dependent complement deposition (ADCD) was also induced by prime vaccination and increased following booster doses at day 56 with higher median fluorescence intensity (MFI), indicating greater complement deposition, observed in recipients of a standard booster dose compared to those of a half dose (Fig. 4c and Supplementary Table 5).
The capacity of the ChAdOx1 nCoV-19 vaccine to induce anti-spike antibody-dependent NK cell activation (ADNKA) in humans was also explored and reported as the capacity to trigger CD107a expression (Fig. 4d). Results demonstrate that single-dose ChAdOx1 nCoV-19 induced low ADNKA responses, which were boosted by the second dose given either at day 28 or day 56. The dose used for boosting at D56 had no impact on the resulting ADNKA measured at D70 (SD/SD 56, median: 5.78, IQR: 4.33–7.7; SD/LD 56, median: 5.29, IQR: 3.61–6.13), whereas ADNKA measured at day 42 after boosting at day 28 was lower (median: 3.96, IQR: 3.44–5.36). The responses observed after two doses of vaccines were within a similar range to those detected in a cohort of 21 convalescent patients with COVID-19 (median: 5.31, IQR: 2.97–8.77), whereas no change was detected after MenACWY vaccination (Supplementary Table 5).
Cellular response
As reported previously17, total antigen-specific T cell responses measured by IFN-γ ELISpot were induced and peaked 14 d after the first vaccination. In ten people who received a booster dose at day 28 (SD/SD D28), responses after a further 28 d following boost vaccination (that is, D56) were the same as in a group of previously reported17 participants (n = 43) who had not received a booster dose at the same time point (P = 0.1126). Similarly, there was no significant difference in the magnitude of spike-specific T cell responses at 28 d following boost vaccination between the 28- or 56-d interval groups (P = 0.736, ANOVA comparing three boosted groups; Fig. 2c and Supplementary Table 6).
Anti-vector immunity
Anti-ChAdOx1 NAbs were induced after the first vaccination and remained elevated above baseline until 84 d after enrollment (last time point assessed). In the groups receiving the second vaccination at day 56, there was a slight decline between day 28 and day 56 followed by a slight boost after the second vaccination, again resulting in similar responses after the first and second vaccinations (P = 0.351) and no differences between groups 28 d after the booster dose, regardless of interval or dose (Supplementary Fig. 3a and Supplementary Table 7). Anti-ChAdOx1 NAb titers at the time of the second dose did not correlate with spike-specific antibody responses following the second vaccination measured by standardized ELISA 28 d after the boost (P = 0.195) or T cell response measured by IFN-γ ELISpot 28 d after the boost dose (P = 0.994), for any vaccination regimen (Supplementary Fig. 3b). Nine participants had positive (>1) anti-ChAdOx1 neutralization titers at baseline. In this small sample, no correlation with anti-ChAdOx1 neutralizing titers at day 28 was evident. Additionally, there was no correlation between preexisting immunity to the ChAdOx1 vector and reactogenicity at second vaccination (Supplementary Fig. 4).
Discussion
We present strong evidence that a second dose of ChAdOx1 nCoV-19 enhances both the titer and the functionality of the antibody response, when measured at early times after the booster dose17. Additionally, we show clear evidence that a booster dose is less reactogenic than the first dose. The data presented here were key to supporting the decision to change from a one- to two-dose regimen for the phase 3 trials of ChAdOx1 nCoV-19, which are now underway. Tolerability of vaccines is important for public acceptance, and the expected reactogenicity profile of the different vaccine products that may be used to control SARS-CoV-2 has to be fully characterized and communicated to future vaccine recipients before successful deployment17. We note that vaccinations were temporarily paused following the triggering of a holding rule in another clinical trial of ChAdOx1 nCoV-19. Vaccinations were resumed in this trial shortly afterward, following review by the independent trial data safety monitoring board (DSMB) and UK regulatory agency, the Medicines and Healthcare products Regulatory Agency. Here we show clearly that, although systemic reactogenicity is prominent after a priming dose of ChAdOx1 nCoV-19, a second dose is consistently less reactogenic, regardless of dose interval. As participants in SD/SD D56 and SD/LD D56 groups were asked to volunteer for a second dose after initially being recruited for one dose only, there was a possibility of selection bias in these groups towards individuals who experienced milder side-effects after the prime vaccination. However, local and systemic reactions seen after priming doses in these groups were comparable to those seen in the ChAdOx1 nCoV-19 participants in the phase 1/2 trial as previously reported17, suggesting this group is representative of the wider trial population. The observation of reduced second-dose reactogenicity is in contrast to reported profiles of two mRNA vaccines for COVID-19 and a protein-adjuvant vaccine technology, in which, although generally well tolerated, reactogenicity increased with the second dose4,18,24. This phenomenon is noteworthy since it is conceivable that additional doses may be required in the future to sustain immunity. Schedules that mix the different vaccine technologies in heterologous prime-boost regimens may maximize immunogenicity, while limiting reactogenicity, and could result in innovative strategies that harness the strengths of the different technologies.
There was no association between reactogenicity and presence or absence of antibodies to either SARS-CoV-2 or ChAdOx1 at the time of vaccination. This is an important finding when considering extended use of the vaccine after licensure, when antibody screening will not be performed before vaccination and a variable proportion of the population will already have been exposed to SARS-CoV-2. Antibodies against the ChAdOx1 vector are induced by the first vaccination but do not prevent boosting and are not further increased by the second vaccination with either a 4-week or 8-week interval. These observations are also important for further development of viral vectors in general, particularly as multiple vaccines are being developed that use the same viral vectors as those in development for SARS-CoV-2 vaccines, including Ad26, Ad5 and ChAdOx12,3,25,26,27.
T cell responses, here measured by IFN-γ ELISpot, were induced rapidly after vaccination and are well maintained in all dosing regimens and intervals between vaccinations. Although NAbs are frequently considered a critical immune response after viral infection, T cells also play important roles during COVID-19. Studies have shown that SARS-CoV-2-specific CD4+ and CD8+ T cells were associated with milder disease in acute and convalescent individuals28. It has previously been shown that during influenza infection, T cells attenuate overt disease and play a key role in disease mitigation29,30,31. Furthermore, an intriguing study in COVID-19 suggests that T cells may correlate with protection32. Additionally, adoptive transfer experiments have demonstrated that coronavirus-specific T cells are sufficient to protect immunodeficient mice from a mouse-adapted SARS strain challenge33, as well as chicks from an infectious bronchitis virus34. IFN-γ ELISpot results demonstrate that, across all regimens, responses are equally well maintained to 28 d after final vaccination irrespective of vaccination interval. Cytotoxic T cells are responsible for destroying virus-infected cells, thus preventing further spread of the virus after infection. The finding that SARS-CoV-2 is capable of cell-to-cell spread35 highlights the importance of this arm of the immune response, as once infection has taken place, the virus is shielded from NAb binding. Helper T cells are also critical to support B cell function for the initial and continued production of antibody. However, the types of T cell responses elicited by this, and other SARS-CoV-2 vaccines, and their roles in protection from infection or disease severity require further investigation.
Correlates of protection against SARS-CoV-2 in humans are currently unknown, but the assumption from animal studies is that NAbs directed against the spike protein may be critically important7,13. Here we show that NAbs are consistently induced across two different dosing intervals, as demonstrated across two assays using either live virus neutralization or pseudotyped virus neutralization as a readout, although we acknowledge that we do not yet have data on the durability of these responses. These observations are similar to those reported for several other COVID-19 vaccines, in which higher titers of NAbs are produced following two-dose vaccination regimens3,4,18,24. As seen previously after adenoviral vector-based vaccination, the data presented here show that IgG subclasses 1 and 3 are induced by ChAdOx1 nCoV-19 (refs. 20,21,22,36,37). Here we see induction of IgA and IgM, in addition to antibody-dependent functional activities (ADMP, ADNP, ADCD and ADNKA), and levels are higher following a second dose.
While the titer of NAbs capable of preventing cellular invasion has emerged as the strongest correlate of protection in preclinical SARS-CoV-2 vaccine studies, non-neutralizing functional activities are increasingly recognized as important mediators of viral control, working in tandem with CD8+ T cells to kill virally infected cells in the host38,39. In preclinical studies of SARS-CoV-2 vaccination, Fc-mediated antibody functions, including ADCD and ADNKA, correlated with protection against infection following viral challenge, and, in combination with NAbs, enhanced the ability to distinguish fully protected rhesus macaques from those which become infected7,8. In this study, ADNP, ADMP and ADNKA responses induced by two doses of ChAdOx1 nCoV-19 were in the same range or higher than that observed in samples from convalescent individuals collected more than 1 month after disease. As shown on the polar plots, broadly similar functional antibody profiles were induced after vaccination, with mixed IgG1, IgG3 and phagocytosis. In contrast to responses in vaccinated individuals, higher titers of anti-spike IgA and IgM antibody isotypes and ADCD were observed in convalescent samples suggesting a qualitative difference in vaccine-induced humoral immunity compared with immune responses following natural infection. One previous study of hospitalized COVID-19 patients demonstrated that the presence of broadly functional anti-spike antibodies in early disease correlated with survival14. This suggests a possible protective effect of Fc-mediated anti-spike antibody effector functions against COVID-19, which, although requiring further investigation as to their role in SARS-CoV-2 immunity, we have now also demonstrated here that they may be induced through prime-boost ChAdOx1 nCoV-19 vaccination.
It has been hypothesized that preexisting anti-vector immunity may affect immune responses induced by human adenoviral vector-based vaccinations and their resulting efficacy2,40. However, several studies have shown that this may be a less substantial concern for other viral vector-based vaccines41,42,43. Furthermore, some evidence suggests the negative impact of preexisting vector immunity can be overcome by booster schedules44. For ChAdOx1, a previous seroprevalence study has demonstrated a low prevalence of anti-ChAdOx1 NAbs in adults in both the United Kingdom and Gambia45, suggesting this is likely to be less of an issue with this vector, although further seroprevalence studies in different populations may be helpful.
Phase 3 trials evaluating safety, immunogenicity and efficacy of ChAdOx1 nCoV-19 after two doses in adults are now underway. If these trials are successful in determining vaccine efficacy, the immune functions induced by vaccination and described here will be evaluated in relation to vaccine efficacy. In summary, ChAdOx1 nCoV-19 is well tolerated in a two-dose regimen and induces multifunctional antibody responses that are enhanced by a booster dose, in addition to T cell responses. These data strongly support evaluation of a two-dose vaccine regimen in phase 3 trials.
Methods
Study design and procedures
As previously described, a phase 1/2 participant-blinded, multicenter, randomized controlled trial is currently underway at five UK sites (NCT04324606)17. We now report further on participants in the same trial that received two doses of vaccine, describing solicited adverse events, serious adverse events and secondary or exploratory humoral and cellular immunology endpoints from these groups.
Healthy volunteers between the ages of 18 and 55 were initially randomized to receive a single dose of either ChAdOx1 nCoV-19 at a standard dose of 5 × 1010 viral particles (vp) or a comparator vaccine in a 1:1 ratio. Randomization lists, using block randomization stratified by study group and study site, were generated by the study statistician (M.V.). Block sizes of two and four were chosen to align with the study group sizes and the sequence of enrollment and varied across study groups. Randomization was performed electronically with full allocation concealment within a secure web platform used for the study (REDCap version 9.5.22; Vanderbilt University). A non-randomized open-label group comprising ten participants was also recruited and received a two-dose schedule of ChAdOx1 nCoV-19 28 d apart at a standard dose of 5 × 1010 vp (SD/SD D28). Following encouraging preliminary NAb data from that group, the trial was amended (22 June 2020) to administer a homologous booster dose at 56 d to a selection of volunteers that had originally been allocated to receive a single dose of vaccine. We selected 62 participants from protocol group 2 (n = 408) that had originally been assigned to receive either a single dose of ChAdOx1 nCoV-19 (n = 52) or MenACWY (n = 10) only and who were also seronegative for SARS-CoV-2 spike antigen at baseline to be boosted at the day-56 time point. Group 2 participants who were seronegative were given the choice to either opt in or decline a boost vaccination. The 52 ChAdOx1 nCoV-19 prime-boost participants were randomized to receive either a standard dose boost (n = 20; SD/SD D56, 5 × 1010 vp) or a dose-sparing half-dose boost (n = 32, SD/LD D56, 2.5 × 1010 vp) of ChAdOx1 nCoV-19 at D56. The ten participants who had been primed with MenACWY were administered a second dose of MenACWY at D56, primarily to maintain blinding of vaccine allocation. Participants in group SD/SD D28 were followed up on days 1, 3, 7, 14, 28, 31, 35, 42 and 56. The 62 volunteers boosted at D56 were followed up on days 28, 56, 70 and 84 from prime vaccination. Laboratory capacity meant that, for some assays, not all samples from each participant could be run at every time point. None of the participants in SD/SD D28, SD/SD D56 and SD/LD D56 groups or in the MenACWY arm received prophylactic paracetamol following either prime or boost vaccination.
Reactogenicity and immunogenicity data from all 30 participants in the trial population that were retrospectively found to be seropositive at baseline against SARS-CoV-2 spike antigen and received ChAdOx1 nCoV-19 (Extended Data Fig. 1; group 1 (n = 2), group 2/4 (n = 27) and group 3 (n = 1)) are also shown.
Safety bloods and exploratory immunology samples were collected for all participants. All volunteers were observed for adverse events for 60 min after prime dose and a minimum of 15 min after the booster vaccination. Self-reported solicited and unsolicited adverse events were recorded via electronic diary for up to 28 d following each vaccine administration.
Written informed consent was obtained from all participants before enrollment. The trial is being conducted according to the principles of Good Clinical Practice and is approved by the South Central Berkshire Research Ethics Committee (20/SC/0145) and the UK regulatory agency (the Medicines and Healthcare products Regulatory Agency). An independent DSMB was appointed before the commencement of recruitment as previously described17. Screening and randomization procedures were as previously described17. Participants with a history of possible but unconfirmed COVID-19 infection or those at high risk of exposure were initially excluded from the trial until SARS-CoV-2 serology became available for screening, from which point only volunteers seropositive at screening were excluded. Despite these steps, several participants were later found to be seropositive to SARS-CoV-2 at the baseline pre-vaccination time point and analyzed as the baseline seropositive ChAdOx1 nCoV-19-recipient group (n = 30).
The trial staff preparing and administering the vaccine were not blinded to treatment allocation; however, the vaccine was prepared out of the participant’s sight and was administered using syringes covered with opaque material to ensure participants remained blinded. Only participants enrolled in SD/SD 28 were not randomized or blinded. Clinical investigators and the laboratory team remained blinded to group allocation.
Convalescent plasma samples from adults (≥18 years) with PCR-positive SARS-CoV-2 infection were obtained from symptomatic patients admitted to hospital or from surveillance on health-care workers who did not have symptomatic infection, as previously described17. These samples were tested using ADNK, ADNP, ADMP and ADCA, and anti-spike antibody isotypes were quantified. Different samples were analyzed across the assays, dependent on sample availability, laboratory capacity and assay-specific requirements. Where multiple longitudinal samples were available for the same participant, only one time point is included in the analyses, and the earliest time point (at least 28 d after initial symptoms) was selected. Pre-pandemic plasma samples were acquired from clinical study participants’ visits undertaken in 2012–2013, which were stored, with individuals’ informed consent, in the Oxford Vaccine Centre Biobank (National Research Ethics Service, 10/H0504/2)
Statistical analysis
Summary statistics are presented by study groups, including medians with IQRs or geometric means (95% CI). A limited number of statistical comparisons are included due to a desire not to overanalyze groups with small sample sizes. For comparisons between groups, independent-samples t-test or ANOVA was applied to log-transformed antibody data. For the analysis of the relationship between anti-vector neutralization titers and anti-spike IgG or T cell responses, an unadjusted linear regression was applied. All analyses were conducted using R version 3.6.1.
Power and sample size calculations were performed for the primary efficacy only, which is not presented here. No specific power calculations were carried out for these immunogenicity subgroups, which are secondary endpoints in the study and mostly descriptive in nature. The results presented here are from an interim analysis of secondary or exploratory outcomes for the described groups of this phase 1/2 randomized control trial (NCT04324606).
Vaccines
ChAdOx1 nCoV-19 consists of a replication-deficient adenovirus viral vector, derived from the simian adenovirus Y25 serotype, that expresses a full-length codon-optimized SARS-CoV-2 spike protein (MN908947) with a leader sequence of tissue plasminogen activator (tPA). The recombinant adenovirus ChAdOx1 nCoV-19 was produced as previously described45. The vaccine supplied for prime doses was formulated and vialed by the Clinical Biomanufacturing Facility, University of Oxford, as previously described17. Booster doses were either supplied by the Clinical Biomanufacturing Facility (volunteers in SD/SD D28) or were manufactured at Advent (volunteers in SD/SD D56 and SD/LD D56). Both products were manufactured to current Good Manufacturing Practice. The licensed comparator meningococcal group ACW135Y conjugate vaccine (MenACWY, NIMENRIX, Pfizer) was administered at the standard dose of 0.5 ml. All vaccines were administered intramuscularly as a single injection into the deltoid muscle.
Meso Scale discovery multiplexed immunoassay
Antigen-specific responses to ChAdOx1 nCoV-19 vaccination and/or natural SARS-CoV-2 infection were measured using an MIA. The MIA was developed and performed by MSD and is described in Folegatti et al.17. Briefly, dried plates coated with SARS-CoV-2 spike protein and RBD were blocked, washed and incubated with samples, reference standards and controls. Internal quality controls (QCs) and reference standard reagents were developed from pooled human serum. Following incubation and washing steps, detection antibody was added (MSD SULFO-TAG anti-human IgG), incubated and plates washed again. MSD GOLD Read Buffer B was added and plates read using a MESO SECTOR S 600. Samples at the lower limit of quantification were set to 2.58 for spike protein and 2.60 for RBD, while samples at the upper limit were set to 320,000 for spike protein and 317,073 for RBD.
Public Health England microneutralization assay
Using a similar method to that described above for the Public Health England microneutralization assay (PHE MNA80; described in Folegatti et al.17), the MNA measures microplaques using the ImmunoSpot S6 Ultra-V Analyzer. Briefly, serum and SARS-CoV-2 (Victoria/01/2020, Doherty Institute) virus mixtures were added to monolayers of virus-susceptible Vero/E6 cells for 1 h before replacement of inoculum with overlay (1% wt/vol CMC in complete media). Following a 24-h incubation, cells were fixed with formaldehyde. Microplaques were detected using a SARS-CoV-2 antibody specific for the SARS-CoV-2 RBD spike protein and a rabbit horseradish peroxidase conjugate. Infected microplaques were detected using TrueBlue Substrate. Resulting counts were analyzed in SoftMax Pro v7.0 software. Data presented for the MNA80 microneutralization assays were normalized to allow comparison across assay runs, including previously published time points.
Monogram Biosciences pseudotype neutralization assay
Nab titers were determined using a lentivirus-based SARS-CoV-2 pseudovirus particle expressing spike protein (MN908947). The pseudotype neutralization CoV nAb assay (PseudoNA) is described by Folegatti et al.17 and based on previously described methodologies using HIV-1 pseudovirions46,47,48. Briefly, heat-inactivated, diluted serum samples were incubated with SARS-CoV-2 pseudotyped virus. Nab titers were determined by creating nine serial threefold dilutions of test samples. Irrelevant pseudotyped virus was used as a control. Following incubation of diluted sera and pseudovirus particle, HEK 293 ACE2-transfected cells were added, plates were incubated and luciferase expression measured. Nab titers were reported as the reciprocal of the serum dilution conferring 50% inhibition (ID50) of pseudovirus infection: % inhibition = 100% − (((relative light units (RLU) (vector + sample + diluent) − RLU (background)) / (RLU (vector + diluent) − RLU (background))) × 100%). SARS-CoV-2 nAb assay positive-and negative-control sera were included on each 96-well assay plate.
Antibody-dependent natural killer cell activation assay
To assess the antigen-specific ADNKA, 96-well Nunc MaxiSorp ELISA plates (Thermo Fisher) were coated with recombinant SARS-CoV-2 spike protein at 2.5 μg ml−1 in carbonate/bicarbonate solution (Sigma-Aldrich) for 16 h at 4 °C. Plates were washed with PBS and blocked with 5% BSA in PBS. Serum and plasma samples were plated undiluted in duplicate. Following incubation for 2 h at 37 °C, the plates were washed and the natural killer NK-92 cell line was subjected to retroviral transduction to express human CD16 (PTA-8836, American Type Culture Collection; described by Binyamin et al.49) and added at 105 cells per well in the presence of brefeldin A (10 μg ml−1; Sigma-Aldrich), Golgi Stop (BD Biosciences) and CD107a (1:20 dilution; PE, clone H4A3, BD Biosciences). A sample of cells was separately stained with CD56 (1:1,000 dilution; BV786, clone NCAM16, BD Biosciences) and CD16 (1:10 dilution; AF594, clone GRM-1, Santa Cruz Biotechnology) to verify consistent expression of CD16. After 5 h of incubation, cells were transferred to V-bottom plates and stained for FACS analysis (Extended Data Fig. 5). Live NK cells were identified by fixable LIVE/DEAD staining (1:500 dilution; R780, BD Biosciences). Cells were fixed and data acquired using a BD Fortessa. Percentages of CD107a+ NK cells relative to control wells with spike protein and blocking buffer only were determined using FlowJo software (version 10.7.1). A pre-pandemic pool of three donors and a pool of six hospitalized SARS-CoV-2 infected individuals were plated in triplicate on each plate, for QC of each assay.
Bead coupling for ADMP and ADNP assays
Red fluorescent (580/605) NeutrAvidin-labeled microspheres (Thermo Fisher, F-8875) were freshly coupled to biotinylated SARS-CoV-2 spike protein for each assay. Spike protein (at a concentration of 0.388 µl ml−1) was coupled to the beads at a 3:1 ratio and incubated for 2 h at 37 °C. Beads were washed twice with 0.1% BSA and diluted 100-fold in 0.1% BSA 10 µl was added to each well in the ADNP and ADMP assays.
Antibody-dependent neutrophil phagocytosis
The ADNP assay was based on a previously described protocol50 with some modifications. Whole donor blood, collected in sodium heparin tubes, was treated with ammonium–chloride–potassium lysing buffer (Thermo Fisher, A1049201) for 5 min followed by centrifugation to collect white blood cells. Cells were washed with DPBS (Sigma, D8537), counted and adjusted to 2.5 × 105 cells per ml in medium consisting of RPMI 1640 medium (Sigma, R5886) supplemented with 100 U ml−1 penicillin–streptomycin (Sigma, P4458) and 20 mmol l−1 l-glutamine (Sigma, G7513).
Serum diluted 100× in RPMI was added to antigen-coupled beads in a 96-well plate and incubated for 2 h at 37 °C. All samples were assayed in duplicate, and each plate contained two QC samples in addition to appropriate negative controls. Wells were washed with DPBS, and 50,000 white blood cells were added to each well followed by a further 1-h incubation at 37 °C. Cells were then stained using a cocktail of mouse anti-human CD3 Alexa Fluor 700 (BD Pharmingen, clone UCHT1, nos. 557943 and 9185576; 1:80 dilution), mouse anti-human CD14 APC Cy7 (BD Pharmingen, clone MΦP9, nos. 557831 and 0044497; 1:80 dilution) and mouse anti-human CD66b Pacific Blue (BioLegend; clone G10F5, nos. 305112 and B285068; 1:80 dilution) and incubated for 15 min at room temperature in the dark. Following washing and fixation using 4% paraformaldehyde (Santa Cruz Biotechnology, SC-281692), the proportion of cells containing fluorescently labeled beads was ascertained using flow cytometry (BD, Fortessa X20).
Data were analyzed with FlowJo (BD; version 10), using a gating strategy to select neutrophils (Extended Data Fig. 6). First, neutrophils were gated based on forward and side scatter. Doublets were excluded in the following two steps. Furthermore, T cells and monocytes were excluded using a double-negative gate for CD3 and CD14. The final neutrophil gate was based on CD66b positivity, after which bead-positive cells were gated. In all cases, there was a clear separation between positive and negative populations. Gates were kept consistent between samples but were checked for each sample.
Normalized phagocytic scores were calculated by multiplying the percentage of bead-positive cells by the MFI of the beads within these cells and normalizing against a QC sample set to 1. As multiple plates were run during an experiment, plates failed if any of the QC sample averages were greater than two standard deviations above the mean of that particular QC across plates. In addition, samples were excluded from further analysis if the replicates showed a coefficient of variation of over 25%. All data are derived from one experiment.
Antibody-dependent monocyte phagocytosis
The ADMP assay was based on a previously described protocol51 with some modifications. Briefly, a human monocytic THP-1 cell line (American Type Culture Collection) was grown and maintained using supplier instructions. Serum was diluted 4,000× in RPMI and was added to antigen-coupled beads in a 96-well plate and incubated for 2 h at 37 °C. All samples were assayed in duplicate, and each plate contained two QC samples in addition to appropriate negative controls. At the end of the 2-h incubation period, wells were washed with RPMI and 25,000 THP-1 cells diluted in medium consisting of RPMI 1640 medium (Sigma, R5886) supplemented with 100 U ml−1 penicillin–streptomycin (Sigma, P4458) and 20 mmol l−1 l-glutamine (Sigma, G7513) were added to each well. Cells were incubated with the antibody-coated beads for 18 h at 37 °C. After the 18-h incubation period, cells were washed with PBS and fixed using 4% paraformaldehyde. The proportion of cells containing fluorescently labeled beads was measured using flow cytometry (BD, Fortessa X20).
Data were analyzed with FlowJo (BD, version 10), using a gating strategy to select the THP-1 cells (Extended Data Fig. 7). First THP-1 cells were gated based on forward and side scatter to exclude debris. Doublets were excluded in the following two steps, after which bead-positive cells were gated. There was a clear separation between the positive and negative population. Gates were kept consistent between samples but were checked for each sample.
Normalized phagocytic scores were calculated by multiplying the percentage of bead-positive cells with the MFI of the beads within these cells and normalizing against a QC sample set to 1. As multiple plates were run during an experiment, plates failed if any of the QC sample averages were greater than two standard deviations above the mean of that particular QC across plates. In addition, samples were excluded from further analysis if the replicates showed a coefficient of variation of over 25%. All data were derived from one experiment.
Antibody-dependent complement deposition
SPHERO carboxyl magnetic blue fluorescent beads (Spherotech) were coupled with SARS-CoV-2 whole spike protein (Lake Pharma, 46328) using a two-step sulpho-NHS/EDC process detailed by Brown et al.52. Spike protein was included at saturation levels and coupling confirmed by the binding of IgG from a COVID-19 convalescent donor known to have high levels of anti-spike protein IgG.
Heat-inactivated test serum (4 µl of 1:10 dilution, in duplicate) was added to 16 µl blocking buffer (PBS, 2% BSA and BB). This was followed by 20 µl of SARS-CoV-2 spike protein-coated magnetic beads (50 beads per µl) to give a final serum dilution of 1:100, and the mixture was incubated at 25 °C for 30 min with shaking at 900 r.p.m. The beads were washed twice in 200 µl wash buffer (BB + 0.05% Tween-20), then resuspended in 50 µl BB containing 10% IgG- and IgM-depleted human plasma (prepared per Lesne et al.53) and incubated at 37 °C for 15 min with shaking at 900 r.p.m. Beads were next washed twice with 200 µl wash buffer and resuspended in 100 µl FITC-conjugated rabbit anti-human C3c polyclonal antibody (Abcam) diluted 1:500 in BB and incubated at room temperature in the dark. After two more washes with 200 µl wash buffer, the samples were resuspended in 60 µl HBSS and analyzed using a CytoFLEX S flow cytometer (Beckman Coulter) and CytExpert software (Extended Data Fig. 8). For each sample, a minimum of 100 beads were collected. Conjugated beads were gated based on forward scatter and violet side scatter and then presented on a histogram showing APC levels. The APC peak was gated and displayed on a FITC histogram, which represents deposition of C3b/iC3b. The MFI of a complement-only, no-serum control was subtracted from each test sample to give the antibody-dependent median FITC fluorescence. Test sera were run in duplicate and data are from a single experiment.
Ex vivo IFN-γ ELISpot
Ex vivo ELISpots were carried out to measure SARS-CoV-2 spike antigen-specific cellular immune responses as previously described17. Briefly, fresh PBMCs were separated from whole blood with lithium heparin by density centrifugation within 4 h of venipuncture. A total of 253 synthetic peptides (15-mers overlapping by ten amino acids) spanning the entire vaccine insert, including the tPA leader sequence, were used to stimulate PBMCs (Pro-Immune). Peptides were pooled into 12 pools for the SARS-CoV-2 spike protein containing 18 to 24 peptides, along with a single pool of 5 peptides for the tPA leader. Peptide sequences and pooling were previously described17. Data were analyzed according to a QC standard operational procedure.
Anti-ChAdOx1 neutralization assay
Chimpanzee adenovirus ChAdOx1-specific NAb titers were assessed using a secreted placental alkaline phosphatase (SEAP) quantification assay as described previously45 with the following minor modifications. Briefly, GripTite 293 MSR cells (Invitrogen, R795-07; cultured per manufacturer’s instructions) were seeded in 96-well plates the previous day at 3 × 104 cells per well. Serial dilutions in 0% FBS–DMEM (phenol red free; GibcoBRL, 31053-028) of heat-inactivated test serum or plasma were mixed at a 1:1 ratio with the ChAdOx1–SEAP reporter vector (at a fixed concentration) and incubated for 1 h to allow antibody binding. Multiple virus/medium-only controls, as well as previously titered serum/plasma controls were included. Serum/plasma-virus mixes were applied to cells for 1 h to allow infection, before replacement with 10% FBS–DMEM (phenol red free) for 24 h. Final serum dilutions were 1:18 to 1:4,608, and each serum sample was tested in duplicate. For each sample, SEAP concentration was assessed in 50 μl aliquots of culture supernatant, with CPSD as an indicator substrate (Tropix Phospha-Light Chemiluminescent Assay Kit; Life Technologies, T1017). Luminescence intensity was measured using a Varioskan Flash luminometer (Thermo Fisher). Serum dilution neutralization titers were measured by linear interpolation of adjacent values (to 50% inhibition) to determine the serum dilution required to reduce SEAP concentration by 50% compared to wells with virus alone, using a standardized spreadsheet.
IgG standardized ELISA
Antigen-specific total IgG was detected with an in-house indirect ELISA using trimeric SARS-CoV-2 spike protein, as described previously17. Briefly, ELISA plates were coated with 2 μg ml−1 spike protein for at least 16 h at 4˚C. After a washing step, diluted serum from vaccinees and relevant controls was added and incubated for 2 h at room temperature. Secondary antibody conjugated to alkaline phosphatase was added after a further washing step. Development was performed using 4-nitrophenyl phosphate in diethanolamine substrate buffer. Optical density was measured at 405 nm and data are presented as standardized EUs.
Isotype and subclass standardized ELISA
Standardized ELISA was performed on donor serum to quantify circulating SARS-CoV-2 spike-specific IgG1, IgG3, IgA and IgM. Nunc MaxiSorp ELISA plates (Thermo Fisher) were coated overnight (≥16 h) at 4 °C with 50 µl per well of 5 µg ml−1 SARS-CoV-2 full-length trimeric spike protein (FL-S) diluted in PBS. This protein was produced as follows. A soluble SARS-CoV-2 FL-S protein (MN908947; Wuhan-Hu-1) construct encoding residues 1–1,213 with two sets of mutations that stabilize the protein in a pre-fusion conformation (removal of a furin cleavage site and the introduction of two proline residues, K983P and V984P) was expressed as described54. The endogenous viral signal peptide was retained at the N terminus (residues 1–14), a C-terminal T4-foldon domain incorporated to promote association of monomers into trimers to reflect the native transmembrane viral protein and a C-terminal His6 tag included for nickel-based affinity purification. FL-S was transiently expressed in Expi293 (Thermo Fisher) and protein purified from culture supernatants by immobilized metal affinity followed by gel filtration in Tris-buffered saline (pH 7.4) buffer.
Plates were washed 3× with 0.05% PBS/Tween (PBS/T) and tapped dry. Plates were blocked for 1 h with 100 µl per well of Blocker Casein in PBS (Thermo Fisher) at 20 °C. Test samples were diluted in blocking buffer (minimum dilution of 1:50), and 50 µl per well was added to the plate in triplicate. For each immunoglobulin isotype or subclass being tested, the respective reference serum (made from a pool of high-titer donor serum) was diluted in blocking buffer in a twofold dilution series to form a ten-point standard curve. Three independent dilutions of the reference serum were made (with a dilution factor corresponding to the fourth point in the standard curve) to serve as internal controls. The standard curve and internal controls were added to the plate at 50 µl per well in duplicate. Plates were incubated for 2 h at 37 °C with 300 r.p.m. shaking and then washed 3× with PBS/T and tapped dry. Secondary antibody was diluted in blocking buffer and 50 µl per well was added. The secondary antibody used was dependent on the immunoglobulin subclass or isotype being detected. These were mouse anti-human IgG1 hinge-AP, mouse anti-human IgG3 hinge-AP, goat anti-human IgA-AP and goat anti-human IgM-AP (Southern Biotech). Plates were incubated for 1 h at 37 °C with 300 r.p.m. shaking. Plates were washed 3× with PBS/T and tapped dry. A total of 100 µl per well of PNPP alkaline phosphatase substrate (Thermo Fisher) was added, and plates were incubated for 1–4 h at 37 °C with 300 r.p.m. shaking. Optical density at 405 nm was measured using an ELx808 absorbance reader (BioTek) until the internal control reached an OD405 of 1. The reciprocal of the internal control dilution giving an OD405 of 1 was used to assign an EU value of the standard. Gen5 ELISA software v3.04 (BioTek) was used to convert the OD405 of test samples into EUs by interpolating from the linear range of the standard curve fitted to a four-parameter logistics model. Any samples with an OD405 below the linear range of the standard curve at the minimum dilution tested were assigned a minimum EU value according to the lower limit of quantification of the assay.
Isotype and subclass OD ELISA
Antigen-specific IgG2 and IgG4 responses were detected in the absence of an antigen-specific serum control. Nunc MaxiSorp ELISA plates (Thermo Fisher) were coated with 50 µl per well of 5 µg ml−1 SARS-CoV-2 trimeric spike protein (The Jenner Institute, University of Oxford). Plates were also coated with a specified concentration of a commercial human immunoglobulin control: recombinant human IgG2 lambda or recombinant human IgG4 lambda (Bio-Rad). Plates were left overnight (≥16 h) at 4 °C. Plates were washed 3× with 0.05% PBS/T and tapped dry. Plates were blocked for 1 h with 100 µl per well of Blocker Casein in PBS (Thermo Fisher) at 20 °C. Test samples and five pre-pandemic negative-control samples were diluted 1:50 in blocking buffer and 50 µl was added to antigen-coated wells in duplicate. Next, 50 µl of blocking buffer was added to immunoglobulin-coated wells and blank wells. Plates were incubated for 2 h at 37 °C with 300 r.p.m. shaking and then washed 3× with PBS/T and tapped dry. Secondary antibody was diluted in blocking buffer and 50 µl per well was added. The secondary antibody used was dependent on the immunoglobulin subclass being detected: mouse anti-human IgG2 Fd-AP or mouse anti-human IgG4 Fc-AP (Southern Biotech). Plates were incubated for 1 h at 37 °C with 300 r.p.m. shaking. Plates were washed 3× with PBS/T and tapped dry. 100 µl per well of PNPP alkaline phosphatase substrate (Thermo Fisher) was added and plates were incubated for 1–4 h at 37 °C with 300 r.p.m. shaking. Optical density at 405 nm was measured using an ELx808 absorbance reader (BioTek) until the immunoglobulin control reached a specified OD405. Negative cutoffs were calculated using the formula: mean + 7.858 × standard deviation of the OD405 readings of the pre-pandemic negative-control serum samples, where 7.858 is the standard deviation multiplier with a 99.9% confidence level for n = 5 controls55.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The University of Oxford is committed to providing access to anonymized data for non-commercial research at the end of the clinical trial, which is currently scheduled to be 1 year after the last participant is enrolled, unless granted an extension. The University of Oxford will collaborate with AstraZeneca for such requests before disclosure.
Change history
06 May 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41591-021-01372-z
References
WHO COVID-19. Draft Landscape of COVID-19 Candidate Vaccines. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (2020).
Zhu, F. C. et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 396, 479–488 (2020).
Logunov, D. Y. et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: two open, non-randomised phase 1/2 studies from Russia. Lancet 396, 887–897 (2020).
Jackson, L. A. et al. An mRNA vaccine against SARS-CoV-2—preliminary report. N. Engl. J. Med. https://doi.org/10.1056/nejmoa2022483 (2020).
Xia, S. et al. Effect of an inactivated vaccine against SARS-CoV-2 on safety and immunogenicity outcomes: interim analysis of 2 randomized clinical trials. JAMA 324, 951–960 (2020).
Xia, S. et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: a randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30831-8 (2020).
Yu, J. et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369, 806–811 (2020).
Mercado, N. B. et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature https://doi.org/10.1038/s41586-020-2607-z (2020).
Robbiani, D. F. et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584, 437–442 (2020).
Wu, F. et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. Preprint at medRxiv https://doi.org/10.1101/2020.03.30.20047365 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Levin, A. T., Cochran, K. B. & Walsh, S. P. Assessing the age specificity of infection fatality rates for COVID-19: meta-analysis & public policy implications. Preprint at medRxiv https://doi.org/10.1101/2020.07.23.20160895 (2020).
Chandrashekar, A. et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science 369, 812–817 (2020).
Atyeo, C. et al. Distinct early serological signatures track with SARS-CoV-2 survival. Immunity 53, 1–9 (2020).
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181, 1489–1501 (2020).
Sekine, T. et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 183, 158–168 (2020).
Folegatti, P. M. et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 396, 467–478 (2020).
Keech, C. et al. Phase 1–2 trial of a SARS-CoV-2 recombinant spike protein nanoparticle vaccine. N. Engl. J. Med. 1–13 https://doi.org/10.1056/nejmoa2026920 (2020).
Walsh, E. E. et al. Safety and immunogenicity of two RNA-Based COVID-19 vaccine candidates. N. Engl. J. Med. 1–13 https://doi.org/10.1056/nejmoa2027906 (2020).
Hodgson, S. H. et al. Combining viral vectored and protein-in-adjuvant vaccines against the blood-stage malaria antigen ama1: report on a phase 1a clinical trial. Mol. Ther. 22, 2142–2154 (2014).
Payne, R. O. et al. Human vaccination against RH5 induces neutralizing antimalarial antibodies that inhibit RH5 invasion complex interactions. JCI Insight 2, 1–19 (2017).
Barouch, D. et al. Evaluation of a mosaic HIV-1 vaccine in a randomized, double-blinded, placebo-controlled phase I/IIa clinical trial and in rhesus monkeys. Lancet 392, 232–243 (2018).
Ewer, K. et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. https://doi.org/10.1038/s41591-020-01194-5 (2021).
Sahin, U. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature https://doi.org/10.1038/s41586-020-2814-7 (2020).
Anywaine, Z. et al. Safety and immunogenicity of a 2-dose heterologous vaccination regimen with Ad26.ZEBOV and MVA-BN-Filo ebola vaccines: 12-month data from a phase 1 randomized clinical trial in Uganda and Tanzania. J. Infect. Dis. 220, 46–56 (2019).
Williams, K. et al. Phase 1 safety and immunogenicity study of a respiratory syncytial virus vaccine with an adenovirus 26 vector encoding prefusion F (Ad26.RSV.preF) in adults aged ≥60 years. J. Infect. Dis. 222, 979–988 (2020).
Sadoff, J. et al. Safety and immunogenicity of the Ad26.COV2.S COVID-19 vaccine candidate: interim results of a phase 1/2a, double-blind, randomized, placebo-controlled trial. Preprint at medRxiv https://doi.org/10.1101/2020.09.23.20199604 (2020).
Rydyznski Moderbacher, C. et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 183, 996–1012 (2020).
Sridhar, S. et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. https://doi.org/10.1038/nm.3350 (2013).
McMichael, A. J., Gotch, F. M., Noble, G. R. & Beare, P. A. S. Cytotoxic T cell immunity to influenza. N. Engl. J. Med. https://doi.org/10.1056/nejm198307073090103 (1983).
Wilkinson, T. M. et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. https://doi.org/10.1038/nm.2612 (2012).
Wyllie, D. et al. SARS-CoV-2 responsive T cell numbers are associated with protection from COVID-19: a prospective cohort study in keyworkers. Preprint at medRxiv https://doi.org/10.1101/2020.11.02.20222778 (2020).
Zhao, J., Zhao, J. & Perlman, S. T Cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. https://doi.org/10.1128/jvi.01049-10 (2010).
Seo, S. H., Pei, J., Briles, W. E., Dzielawa, J. & Collisson, E. W. Adoptive transfer of infectious bronchitis virus primed αβ T cells bearing CD8 antigen protects chicks from acute infection. Virology https://doi.org/10.1006/viro.2000.0211 (2000).
Fenrich, M. et al. SARS-CoV-2 dissemination through peripheral nerves explains multiple organ injury. Front. Cell. Neurosci. 14, 1–12 (2020).
Payne, R. O. et al. Human vaccination against Plasmodium vivax Duffy-binding protein induces strain-transcending antibodies. JCI Insight 2, e93683 (2017).
Biswas, S. et al. Assessment of humoral immune responses to blood-stage malaria antigens following ChAd63-MVA immunization, controlled human malaria infection and natural exposure. PLoS ONE 9, e107903 (2014).
Excler, J. L., Ake, J., Robb, M. L., Kim, J. H. & Plotkin, S. A. Nonneutralizing functional antibodies: a new ‘old’ paradigm for HIV vaccines. Clin. Vaccine Immunol. 21, 1023–1036 (2014).
DiLillo, D., Tan, G., Palese, P. & Ravetch, J. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med. 20, 143–151 (2014).
Buchbinder, S. P. et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet https://doi.org/10.1016/S0140-6736(08)61591-3 (2008).
Koch, T. et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: an open-label, phase 1 trial. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30248-6 (2020).
Reisinger, E. C. et al. Immunogenicity, safety and tolerability of the measles-vectored chikungunya virus vaccine MV-CHIK: a double-blind, randomised, placebo-controlled and active-controlled phase 2 trial. Lancet https://doi.org/10.1016/S0140-6736(18)32488-7 (2018).
Ledgerwood, J. E. et al. Chimpanzee adenovirus vector Ebola vaccine. N. Engl. J. Med. https://doi.org/10.1056/nejmoa1410863 (2017).
Li, J. X. et al. Immunity duration of a recombinant adenovirus type-5 vector-based Ebola vaccine and a homologous prime-boost immunisation in healthy adults in China: final report of a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Glob. Health https://doi.org/10.1016/S2214-109X(16)30367-9 (2017).
Dicks, M. D. J. et al. A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. PLoS ONE 7, e40385 (2012).
Petropoulos, C. J. et al. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 44, 920–928 (2000).
Richman, D. D., Wrin, T., Little, S. J. & Petropoulos, C. J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl Acad. Sci. USA 100, 4144–4149 (2003).
Whitcomb, J. M. et al. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob. Agents Chemother. 51, 566–575 (2007).
Binyamin, L. et al. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 180, 6392–6401 (2008).
Karsten, C. B. et al. A versatile high throughput assay to characterize antibody-mediated neutrophil phagocytosis. J. Immunol. Methods 471, 46–56 (2019).
Ackerman, M. E. et al. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J. Immunol. Methods 366, 8–19 (2011).
Brown, E. P. et al. High-throughput, multiplexed IgG subclassing of antigen-specific antibodies from clinical samples. J. Immunol. Methods 386, 117–123 (2012).
Lesne, E. et al. Acellular pertussis vaccines induce anti-pertactin bactericidal antibodies which drives the emergence of pertactin-negative strains. Front. Microbiol. 11, 2108 (2020).
Amanat, F. et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 26, 1033–1036 (2020).
Frey, A., Di, J. & Zurakowski, D. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 221, 35–41 (1998).
Acknowledgements
The funder had no role in the design, execution or analysis of this study. This report is independent research funded by the National Institute for Health Research (NIHR). The views expressed in this publication are those of the authors and not necessarily those of the NIHR or the UK Department of Health and Social Care (DHSC). Additional resources for study delivery were provided by NIHR Southampton Clinical Research Facility and NIHR Southampton Biomedical Research Centre; University Hospital Southampton NHS Foundation Trust; the NIHR Imperial Clinical Research Facility; and NIHR North West London, South London, Wessex and West of England Local Clinical Research Networks; and NIHR Oxford Health Biomedical Research Centre. P.M.F. received funding from the Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior, Brazil (finance code 001). The control vaccine was provided free of charge by the UK DHSC. The authors are grateful to the volunteers who participated in this study. The investigators express their gratitude for the contribution of all the trial participants, the invaluable advice of the international DSMB (Supplementary Information) and the independent members of the trial steering committee. We are grateful for the advice and intellectual support from K.S. Campbell (Institute for Cancer Research, Philadelphia) with the NK cell line culture and use. The authors are grateful to the senior management at AstraZeneca for facilitating and funding the manufacture of the AZD1222 vaccine candidate, the pseudovirus neutralization assays and Meso Scale antibody assay used in this study. AstraZeneca reviewed the data from the study and the final manuscript before submission, but the authors retained editorial control. Finally, we acknowledge UKRI, NIHR, CEPI, NIHR Oxford Biomedical Research Centre, Thames Valley and South Midland’s NIHR Clinical Research Network and AstraZeneca.
Author information
Authors and Affiliations
Consortia
Contributions
S.C.G. and A.J.P. conceived the trial and A.J.P. is the chief investigator. A.J.P., P.M.F., D.J., H.R. and M.V. contributed to the protocol and design of the study. A.V.S.H. is the study site principal investigator. J.B., S.B.R., C.D., K.E., C.G., R.H., J.H., L.S., M.V., D.B., S.B., M.B., M.C., B.C., E.C., N.E., A.F., A.G., B.H., A.L., R.M., J.M., C.R., K.T., D.W. and T.L. were responsible for laboratory testing and assay development. P.K.A., D.J., H.R., P.M.F., A.M.M., M.R., B.A., M.F., S.K., A.L., M. Madhavan., M. Moore., Y.M., E.P., I.P., R.S., M.W. and M.S. contributed to the implementation of the study and/or data collection. M.V. and X.L. conducted the statistical analysis. C.G., A.D.D. and R.T. were responsible for vaccine manufacturing. T.L., M.V., S.C.G., A.J.P., D.J., P.M.F., C.R. and L.S. contributed to the preparation of the report. All authors critically reviewed and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The University of Oxford has entered into a partnership with AstraZeneca for further development of ChAdOx1 nCoV-19. S.C.G. is cofounder of Vaccitech (collaborators in the early development of this vaccine candidate) and named as an inventor on a patent covering use of ChAdOx1 vector-based vaccines and a patent application covering this SARS-CoV-2 vaccine. T.L. is named as an inventor on a patent application covering this SARS-CoV-2 vaccine and was a consultant to Vaccitech for an unrelated project. P.M.F. is a consultant to Vaccitech. A.J.P. is Chair of the UK DHSC Joint Committee on Vaccination & Immunisation but does not participate in discussions on COVID-19 vaccines, and is a member of the Strategic Advisory Group of Experts on Immunization to the WHO (World Health Organization). A.J.P. is an NIHR senior investigator. The views expressed in this article do not necessarily represent the views of the DHSC, Joint Committee on Vaccination & Immunisation, NIHR or WHO. A.V.S.H. reports personal fees from Vaccitech, outside the submitted work, and has a patent ChAdOx1 licensed to Vaccitech, and may benefit from royalty income to the University of Oxford from sales of this vaccine by AstraZeneca and sublicensees. M.S. reports grants from the NIHR; nonfinancial support from AstraZeneca; grants from Janssen, GlaxoSmithKline, MedImmune and Novavax; grants and nonfinancial support from Pfizer; and grants from MCM, outside the submitted work. C.G. reports personal fees from the Duke Human Vaccine Institute, outside the submitted work. A.D.D. reports grants and personal fees from AstraZeneca, outside the submitted work. In addition, A.D.D. has a patent manufacturing process for ChAdOx vectors with royalties paid to AstraZeneca and a patent ChAdOx2 vector with royalties paid to AstraZeneca. The other authors declare no competing interests.
Additional information
Peer review information Alison Farrell was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CONSORT diagram.
A CONSORT diagram showing trial groups and participant flow in this study (NCT04324606).
Extended Data Fig. 2 Solicited local and systemic reactions by baseline seropositivity to SARS-CoV-2 spike protein.
Top panel: Local solicited reactions. Bottom panel: Systemic solicited reactions. Day 0 is the day of vaccination. P = post-vaccination observation period in the clinic. Fever = Mild: 38.0oC to <38.5oC; moderate: 38.5oC to <39.0oC; severe: ≥39.0oC. Feverish = Self-reported feeling of feverishness.
Extended Data Fig. 3 Anti-vector immunity.
Participants received a prime dose of ChAdOx1-nCoV at day 0 and a matched booster dose at either day 28 (SD/SD D28 n = 10) or at day 56 (SD/SD D56 n = 20, SD/LD D56 n = 32). Another group received MenACWY prime followed by D56 boost vaccination (MenACWY n = 10). a, Anti-ChAdOx1 neutralisation titres in three prime-boost groups and in those receiving control vaccine. Median with interquartile range of anti-ChAdOx1 Nab titres, at days 0, 28, 58 and 84 are shown. Values below the limit of detection were assigned a value of 1. (MenACWY n = 10 all timepoints; SD/SD D28 n = 10 at D0, D28, D56, not performed at D70, D84; SD/SD D56 D0 n = 20, D28 n = 19, D56 n = 20, D70 n = 20, D84 n = 19; SD/SD D56 D0 n = 32, D28 n = 32, D56 n = 32, D70 n = 31, D84 n = 31) b, Anti-ChAdOx1 Nab titre at day of boost vs standardised ELISA against SARS-COV-2 spike 28 days post-boost. Anti-ChAdOx1 neutralisation titre at the time of the 2nd dose are not correlated with standardised ELISA output 28 days after the boost (p = 0.844 from linear regression) (SD/SD D28 n = 10, SD/SD D56 n = 20, SD/LD D56 n = 31) c Anti-ChAdOx1 Nab titres at day of boost vs SARS-CoV-2 spike specific T cells measured by IFNγ ELISPOT on day 28 post-boost. Anti-ChAdOx1 neutralisation titre at the time of the 2nd dose are not correlated with IFN-g ELISpots (p = 0.370 from linear regression). (SD/SD D28 n = 10, SD/SD D56 n = 20, SD/LD D56 n = 31). Confidence bands show 95% confidence intervals from an unadjusted linear regression of anti-vector neutralisation titres against logged (b) ELISA or (c) ELISpot responses.
Extended Data Fig. 4 Solicited local and systemic reactions at second dose of vaccination by pre-existing immunity to the ChAdOx1 vector.
Top panel: Local solicited reactions. Bottom panel: Systemic solicited reactions. Day 0 is the day of vaccination. P = post-vaccination observation period in the clinic. Fever = Mild: 38.0oC to <38.5oC; moderate: 38.5oC to <39.0oC; severe: ≥39.0oC. Feverish = Self-reported feeling of feverishness.
Extended Data Fig. 5 Example of ADNKA gating strategy.
Viable NK-92® expressing human CD16 were identified by FSC-A vs SSC-A gating, followed by GFP expression, and Live/Dead stain. CD107a expression was then determined from this gated population, an unstimulated sample is shown in the top right panel, and stimulation with a convalescent sample is shown on the bottom right panel.
Extended Data Fig. 6 Example of ADNP gating strategy.
First neutrophils are gated based on forward and side scatter. Doublets are excluded in the following two steps. Furthermore, T cells and monocytes are excluded using a double-negative gate for CD3 and CD14. The final neutrophil gate is based on CD66-positivity, after which bead+ cells are gated. Phagocytic score is calculated by multiplying the percentage of bead-positive cells with the mean fluorescence intensity (MFI) of the beads within these cells to account for the number of beads per cell.
Extended Data Fig. 7 Example of ADMP gating strategy.
First THP-1 cells are gated based on forward and side scatter. Doublets are excluded in the following two steps, after which bead+ cells are gated. Phagocytic score is calculated by multiplying the percentage of bead-positive cells with the mean fluorescence intensity (MFI) of the beads within these cells to account for the number of beads per cell.
Extended Data Fig. 8 Example of ADCD gating strategy.
Example of ADCD gating strategy: Conjugated beads are gated based on Forward Scatter and Violet Side Scatter then presented on an APC-histogram. The APC peak is gated and displayed on a FITC-histogram which represents deposition of C3b/iC3b. The median fluorescence intensity (MFI) of the complement only, no antibody control (complement only) is subtracted from the test sample (sample) to give the antibody-dependent MFI.
Supplementary information
Supplementary Information
Supplementary Tables 1–7, Supplementary Acknowledgements and Study Protocol.
Rights and permissions
About this article

Cite this article
Barrett, J.R., Belij-Rammerstorfer, S., Dold, C. et al. Phase 1/2 trial of SARS-CoV-2 vaccine ChAdOx1 nCoV-19 with a booster dose induces multifunctional antibody responses. Nat Med 27, 279–288 (2021). https://doi.org/10.1038/s41591-020-01179-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-020-01179-4
This article is cited by
-
MVA-based vaccine candidates encoding the native or prefusion-stabilized SARS-CoV-2 spike reveal differential immunogenicity in humans
npj Vaccines (2024)
-
Caveats of chimpanzee ChAdOx1 adenovirus-vectored vaccines to boost anti-SARS-CoV-2 protective immunity in mice
Applied Microbiology and Biotechnology (2024)
-
Impact of timing and combination of different BNT162b2 and ChAdOx1-S COVID-19 basic and booster vaccinations on humoral immunogenicity and reactogenicity in adults
Scientific Reports (2023)
-
Robust induction of functional humoral response by a plant-derived Coronavirus-like particle vaccine candidate for COVID-19
npj Vaccines (2023)
-
Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection
Nature Reviews Immunology (2023)
