
Abstract
De novo and inherited rare genetic disorders (RGDs) are a major cause of human morbidity, frequently involving neuropsychiatric symptoms. Recent advances in genomic technologies and data sharing have revolutionized the identification and diagnosis of RGDs, presenting an opportunity to elucidate the mechanisms underlying neuropsychiatric disorders by investigating the pathophysiology of high-penetrance genetic risk factors. Here we seek out the best path forward for achieving these goals. We think future research will require consistent approaches across multiple RGDs and developmental stages, involving both the characterization of shared neuropsychiatric dimensions in humans and the identification of neurobiological commonalities in model systems. A coordinated and concerted effort across patients, families, researchers, clinicians and institutions, including rapid and broad sharing of data, is now needed to translate these discoveries into urgently needed therapies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Debbie Maizels/Springer Nature.

Debbie Maizels/Springer Nature.

Debbie Maizels/Springer Nature.

Debbie Maizels/Springer Nature.
Similar content being viewed by others


References
US Food and Drug Administration. Orphan Drug Act. (1983).
Loane, M. et al. Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: impact of maternal age and prenatal screening. Eur. J. Hum. Genet. 21, 27–33 (2013).
McKusick-Nathans Institute of Genetic Medicine. Online Mendelian Inheritance in Man, OMIM® (Johns Hopkins University, Baltimore, MD, USA) https://omim.org/ (accessed 28 April 2018).
McRae, J. F. et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017).
Sanders, S. J. et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015).
Catterall, W. A., Kalume, F. & Oakley, J. C. NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859 (2010).
Escayg, A. et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24, 343–345 (2000).
Marshall, C. R. et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017).
Lukowski, A. F., Milojevich, H. M. & Eales, L. Cognitive functioning in children with down syndrome: current knowledge and future directions. Adv. Child Dev. Behav. 56, 257–289 (2019).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012).
Ben-Shalom, R. et al. Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82, 224–232 (2017).
Power, R. A. et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry 70, 22–30 (2013).
Stefansson, H. et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 (2014).
Sanders, S. J. et al. Whole genome sequencing in psychiatric disorders: the WGSPD consortium. Nat. Neurosci. 20, 1661–1668 (2017).
Reuter, M. S. et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 74, 293–299 (2017).
Schaefer, G. B. et al. Array comparative genomic hybridization findings in a cohort referred for an autism evaluation. J. Child Neurol. 25, 1498–1503 (2010).
Retterer, K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016).
Sawyer, S. L. et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 89, 275–284 (2016).
Tammimies, K. et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903 (2015).
Kosmicki, J. A. et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510 (2017).
Weiner, D. J. et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49, 978–985 (2017).
EuroEPINOMICS-RES Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370 (2014).
Heyne, H. O. et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 50, 1048–1053 (2018).
Ganna, A. et al. Ultra-rare disruptive and damaging mutations influence educational attainment in the general population. Nat. Neurosci. 19, 1563–1565 (2016).
Singh, T. et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 49, 1167–1173 (2017).
Willsey, A. J. et al. De Novo coding variants are strongly associated with Tourette disorder. Neuron 94, 486–499.e9 (2017).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014).
Purcell, S. M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014).
Finkel, R. S. et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Schneider, M. et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 171, 627–639 (2014).
Rees, E. et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol. Psychiatry 19, 37–40 (2014).
Kendall, K. M. et al. Archival report cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK Biobank subjects. Biol. Psychiatry 83, 103–110 (2016).
D’Angelo, D. et al. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 73, 20–30 (2016).
Niemi, M. E. K. et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271 (2018).
Moreno-De-Luca, A. et al. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry 72, 119–126 (2015).
Insel, T. R. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am. J. Psychiatry 171, 395–397 (2014).
Cuthbert, B. N. & Insel, T. R. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med. 11, 126 (2013).
Cuthbert, B. N. Research Domain Criteria: toward future psychiatric nosologies. Dialog-. Clin. Neurosci. 17, 89–97 (2015).
Constantino, J. N. et al. Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview-revised. J. Autism Dev. Disord. 33, 427–433 (2003).
van Os, J. & Reininghaus, U. Psychosis as a transdiagnostic and extended phenotype in the general population. World Psychiatry 15, 118–124 (2016).
Olsen, L. et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry 5, 573–580 (2018).
Männik, K. et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA. 313, 2044–2054 (2015).
Simons Vip, C., Spiro, J. E. & Chung, W. K. Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron 73, 1063–1067 (2012).
Stessman, H. A., Bernier, R. & Eichler, E. E. A genotype-first approach to defining the subtypes of a complex disease. Cell 156, 872–877 (2014).
He, X. et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 9, e1003671 (2013).
Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014).
Singh, T. et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016).
Ahn, K. et al. High rate of disease-related copy number variations in childhood onset schizophrenia. Mol. Psychiatry 19, 568–572 (2014).
Satterstrom, F.K. et al. Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. Preprint at https://www.biorxiv.org/content/10.1101/484113v3 (2018).
Mighell, T. L., Evans-Dutson, S. & O’Roak, B. J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotypes relationships. Am. J. Hum. Genet. 102, 943–955 (2018).
Huguet, G. et al. Measuring and estimating the effect sizes of copy number variants on general intelligence in community-based samples. JAMA Psychiatry 75, 447–457 (2018).
Geschwind, D. H. Autism: many genes, common pathways? Cell 135, 391–395 (2008).
Cheng, H. et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 28, 2900–2919 (2019).
Chakravarti, A., Clark, A. G. & Mootha, V. K. Distilling pathophysiology from complex disease genetics. Cell 155, 21–26 (2013).
Gandal, M. J., Leppa, V., Won, H., Parikshak, N. N. & Geschwind, D. H. The road to precision psychiatry: translating genetics into disease mechanisms. Nat. Neurosci. 19, 1397–1407 (2016).
Parikshak, N. N., Gandal, M. J. & Geschwind, D. H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 16, 441–458 (2015).
Amin, N. D. & Paşca, S. P. Building models of brain disorders with three-dimensional organoids. Neuron 100, 389–405 (2018).
Sun, Y. et al. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. eLife 5, e13073 (2016).
Willsey, A. J. et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013).
Parikshak, N. N. et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013).
Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019).
Cargnin, F. et al. FOXG1 orchestrates neocortical organization and cortico-cortical connections. Neuron 100, 1083–1096.e5 (2018).
Guloksuz, S., Pries, L. K. & van Os, J. Application of network methods for understanding mental disorders: pitfalls and promise. Psychol. Med. 47, 2743–2752 (2017).
Sheffield, J. M. et al. Transdiagnostic associations between functional brain network integrity and cognition. JAMA Psychiatry 74, 605–613 (2017).
Cao, H. et al. Toward leveraging human connectomic data in large consortia. Generalizability of fMRI-based brain graphs across sites, sessions, and paradigms. Cereb. Cortex (2018).
Anticevic, A. et al. Association of Thalamic dysconnectivity and conversion to psychosis in youth and young adults at elevated clinical risk. JAMA Psychiatry 72, 882–891 (2015).
Bruno, J. L. et al. Longitudinal identification of clinically distinct neurophenotypes in young children with fragile X syndrome. Proc. Natl. Acad. Sci. USA 114, 10767–10772 (2017).
Hazlett, H. C. et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351 (2017).
Bearden, C. E. & Thompson, P. M. Emerging global initiatives in neurogenetics: the Enhancing Neuroimaging Genetics through Meta-analysis (ENIGMA) consortium. Neuron 94, 232–236 (2017).
Thompson, P. M. et al. ENIGMA and the individual: predicting factors that affect the brain in 35 countries worldwide. Neuroimage 145 Pt B, 389–408 (2017).
Thompson, P. M. et al. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 8, 153–182 (2014).
National Advisory Mental Health Council Workgroup on Genomics. Opportunities and Challenges of Psychiatric Genetics (NAHMC, 2018).
Skene, N. G. et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 50, 825–833 (2018).
Deisseroth, K., Etkin, A. & Malenka, R. C. Optogenetics and the circuit dynamics of psychiatric disease. J. Am. Med. Assoc. 313, 2019–2020 (2015).
Stoodley, C. J. et al. Author Correction: Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat. Neurosci. 21, 1016 (2018).
Anthony, T. E. et al. Control of stress-induced persistent anxiety by an extra-amygdala septohypothalamic circuit. Cell 156, 522–536 (2014).
Stewart, A. M. & Kalueff, A. V. Developing better and more valid animal models of brain disorders. Behav. Brain Res. 276, 28–31 (2015).
LeBlanc, J. J. et al. Visual evoked potentials detect cortical processing deficits in Rett syndrome. Ann. Neurol. 78, 775–786 (2015).
Lovelace, J. W., Ethell, I. M., Binder, D. K. & Razak, K. A. Translation-relevant EEG phenotypes in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 115, 39–48 (2018).
Chadman, K. K., Yang, M. & Crawley, J. N. Criteria for validating mouse models of psychiatric diseases. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 150B, 1–11 (2009).
Galvão-Coelho, N. L., Galvão, A. C. M., da Silva, F. S. & de Sousa, M. B. C. Common marmosets: a potential translational animal model of juvenile depression. Front. Psychiatry 8, 175 (2017).
Oikonomidis, L. et al. A dimensional approach to modeling symptoms of neuropsychiatric disorders in the marmoset monkey. Dev. Neurobiol. 77, 328–353 (2017).
Mao, P., Cui, D., Zhao, X.-D. & Ma, Y.-Y. Prefrontal dysfunction and a monkey model of schizophrenia. Neurosci. Bull. 31, 235–241 (2015).
Kotani, M. et al. The atypical antipsychotic blonanserin reverses (+)-PD-128907- and ketamine-induced deficit in executive function in common marmosets. Behav. Brain Res. 305, 212–217 (2016).
Clarke, H. F. et al. Orbitofrontal dopamine depletion upregulates caudate dopamine and alters behavior via changes in reinforcement sensitivity. J. Neurosci. 34, 7663–7676 (2014).
Zhou, Y. et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 570, 326–331 (2019).
Pașca, S. P. The rise of three-dimensional human brain cultures. Nature 553, 437–445 (2018).
Bredenoord, A. L., Clevers, H. & Knoblich, J. A. Human tissues in a dish: The research and ethical implications of organoid technology. Science 355, eaaf9414 (2017).
Birey, F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017).
Wang, J. et al. A resting EEG study of neocortical hyperexcitability and altered functional connectivity in fragile X syndrome. J. Neurodev. Disord. 9, 11 (2017).
Sahin, M. et al. Discovering translational biomarkers in neurodevelopmental disorders. Nat. Rev. Drug Discov. 18, 235–236 (2018).
Donaldson, Z. R. & Hen, R. From psychiatric disorders to animal models: a bidirectional and dimensional approach. Biol. Psychiatry 77, 15–21 (2015).
Spencer, C. M. et al. Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res. 4, 40–56 (2011).
Aylor, D. L. et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 21, 1213–1222 (2011).
Vockley, J. et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet. Med. 16, 188–200 (2014).
Berry-Kravis, E. M. et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 17, 280–299 (2018).
Krueger, D. A. et al. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 4, 877–887 (2017).
O’Leary, H. M. et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann. Clin. Transl. Neurol. 5, 323–332 (2018).
Guy, J., Gan, J., Selfridge, J., Cobb, S. & Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147 (2007).
Henderson, C. et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4, 152ra128 (2012).
Dolan, B. M. et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 110, 5671–5676 (2013).
Jacquemont, S. et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 478, 97–102 (2011).
Wiesel, T. N. & Hubel, D. H. Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017 (1963).
Berry-Kravis, E. AFQ056 for language learning in children with FXS. https://clinicaltrials.gov/ct2/show/NCT02920892.
Bebin, M. Preventing epilepsy using vigabatrin in infants with tuberous sclerosis complex. https://clinicaltrials.gov/ct2/show/NCT02849457.
Jozwiak, S. Long-term, prospective study evaluating clinical and molecular biomarkers of epileptogenesis in a genetic model of epilepsy—Tuberous Sclerosis Complex (EPISTOP). https://clinicaltrials.gov/ct2/show/NCT02098759.
Kothari, C. et al. Phelan-McDermid syndrome data network: integrating patient reported outcomes with clinical notes and curated genetic reports. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 177, 613–624 (2018).
Kohane, I. S. Using electronic health records to drive discovery in disease genomics. Nat. Rev. Genet. 12, 417–428 (2011).
Berry-Kravis, E. et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 8, 321ra5 (2016).
van der Vaart, T., Overwater, I. E., Oostenbrink, R., Moll, H. A. & Elgersma, Y. Treatment of cognitive deficits in genetic disorders: a systematic review of clinical trials of diet and drug treatments. JAMA Neurol. 72, 1052–1060 (2015).
Wolff, M. et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336 (2017).
Guerrini, R. & Falchi, M. Dravet syndrome and SCN1A gene mutation related-epilepsies: cognitive impairment and its determinants. Dev. Med. Child Neurol. 53 Suppl 2, 11–15 (2011).
Moreno-De-Luca, D., Moreno-De-Luca, A., Cubells, J. F. & Sanders, S. J. Cross-disorder comparison of four neuropsychiatric CNV loci. Curr. Genet. Med. Rep. 2, 151–161 (2014).
Demkow, U. & Wolańczyk, T. Genetic tests in major psychiatric disorders-integrating molecular medicine with clinical psychiatry—why is it so difficult? Transl. Psychiatry 7, e1151 (2017).
US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER) & Center for Biologics Evaluation and Research (CBER). Rare diseases: common issues in drug development guidance for industry. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM458485.pdf (2019).
Collins, R. What makes UK Biobank special? Lancet 379, 1173–1174 (2012).
Senthil, G., Dutka, T., Bingaman, L. & Lehner, T. Genomic resources for the study of neuropsychiatric disorders. Mol. Psychiatry 22, 1659–1663 (2017).
Bastarache, L. et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 359, 1233–1239 (2018).
Pedersen, C. B. et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol. Psychiatry 23, 6–14 (2018).
Rusk, N. The UK Biobank. Nat. Methods 15, 1001 (2018).
An, J.-Y. & Sanders, S. J. Appreciating the population-wide impact of copy number variants on cognition. Biol. Psychiatry 82, 78–80 (2017).
Köhler, S. et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 45 D1, D865–D876 (2017).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Finucane, B.M. et al. 15q duplication syndrome and related disorders. in Gene Reviews (eds. Pagon, R.A. et al.) (University of Washington, Seattle) https://www.ncbi.nlm.nih.gov/books/NBK367946/ (2016).
Miller, I.O. & Sotero de Menezes, M.A. SCN1A seizure disorders. in Gene Reviews (eds. Pagon, R.A. et al.) (University of Washington, Seattle) https://www.ncbi.nlm.nih.gov/books/NBK1318/ (2007).
Kirov, G. et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry 75, 378–385 (2014).
Moreno-De-Luca, D. et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 18, 1090–1095 (2013).
Malhotra, D. & Sebat, J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241 (2012).
Sanders, S. J. et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 41, 442–456 (2018).
Joseph, L. et al. Characterization of autism spectrum disorder and neurodevelopmental profiles in youth with XYY syndrome. J. Neurodev. Disord. 10, 30 (2018).
Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 107, 961–968 (2010).
Scheldeman, C. et al. mTOR-related neuropathology in mutant tsc2 zebrafish: Phenotypic, transcriptomic and pharmacological analysis. Neurobiol. Dis. 108, 225–237 (2017).
Kelly, E. et al. mGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology 43, 1457–1465 (2018).
Shukla, G. et al. Magnetoencephalographic identification of epileptic focus in children with generalized electroencephalographic (EEG) Features but focal imaging abnormalities. J. Child Neurol. 32, 981–995 (2017).
Pietri, T. et al. The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 7, 118 (2013).
Wu, Y. et al. Characterization of Rett Syndrome-like phenotypes in Mecp2-knockout rats. J. Neurodev. Disord. 8, 23 (2016).
Chen, Y. et al. Modeling Rett syndrome using TALEN-edited MECP2 mutant cynomolgus monkeys. Cell 169, 945–955.e10 (2017).
Pidcock, F. S. et al. Functional outcomes in Rett syndrome. Brain Dev. 38, 76–81 (2016).
Acknowledgements
This paper offers a synthesis of the ideas generated at the NIMH-sponsored workshop “Rare Genetic Disease Workshop: Window into Genomic Risk and Resilience of Mental Disorders,” held in September 2017, with the goal of discussing research and clinical opportunities presented by recent discoveries of RGDs with high risk for developmental neuropsychiatric disorders. Analyses utilize data generated by the Saguenay Youth Study, OMIM (https://www.omim.org), ExAC (http://exac.broadinstitute.org/) and the DECIPHER Consortium, including the Developmental Disorders Genotype-Phenotype Database (DDG2P, https://decipher.sanger.ac.uk/info/ddg2p). A full list of centers that contributed to the generation of the DECIPHER data is available at http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. We also thank G. Senthil for helpful feedback on the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Hannah Stower was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
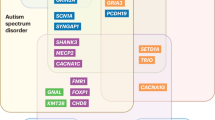
Extended Data Fig. 1 Impact of RGDs on neuropsychiatric domains.
a, Many RGDs impact cognition, measured by IQ. For CNVs, the decrease in IQ (x axis) can be predicted by considering the pLI score of the genes within the CNV. CNVs that are predicted to markedly reduce IQ are more likely to be de novo (y axis), based on logistic regression (blue line) of 2,743 CNVs detected in patients with neurodevelopmental disorders and the general population (gray distributions at top and bottom). Updated analysis from ref. 54. b, In Fig. 2, we show the odds ratio for ID/NDD, ASD and SCZ across different CNV loci. Here, we show an equivalent plot for single-gene RGDs. Insufficient control data exist to estimate odds ratio, and therefore we show the percentage of cases with ID/NDD, ASD, and IE based on curated publication review applied equally across genes (https://dbd.geisingeradmi.org) with the number of cases are shown in parentheses (see Supplementary Table 2 for numbers). Abbreviations: ID, intellectual disability; NDD, neurodevelopmental delay; ASD, autism spectrum disorder; SCZ, schizophrenia; IE, infantile epilepsy; pLI, probability loss-of-function intolerant.
Extended Data Fig. 2 Thresholds for genome-wide significant association with de novo PTVs.
a, Gene mutability is a function of gene length (cDNA) and sequence context (particularly GC content). b, RGD gene discovery from exome sequencing has been driven by de novo mutations, leading to a bias towards larger genes with higher mutability. c, Thresholds of statistical association (colored lines) are estimated for a given number of de novo PTV mutations (3, 5, 10, and 20) as cohort size (x axis) and gene mutability/size (y axis) varies. P values are estimated based on the rate of de novo PTV mutations in controls4 and a Poisson distribution (see Methods for details). Abbreviations: pLI, probability of loss-of-function intolerance; ASD, autism spectrum disorder; DDD: Deciphering Developmental Disorders; GC content, guanine-cytosine content.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary Tables 3 and 5.
Supplementary Table
Supplementary Tables 1, 2, and 4
Rights and permissions
About this article

Cite this article
Sanders, S.J., Sahin, M., Hostyk, J. et al. A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat Med 25, 1477–1487 (2019). https://doi.org/10.1038/s41591-019-0581-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-019-0581-5
This article is cited by
-
Comparing ability and norm-referenced scores as clinical trial outcomes for neurodevelopmental disabilities: a simulation study
Journal of Neurodevelopmental Disorders (2023)
-
Deep phenotypic analysis of psychiatric features in genetically defined cohorts: application to XYY syndrome
Journal of Neurodevelopmental Disorders (2023)
-
A genetics-first approach to understanding autism and schizophrenia spectrum disorders: the 22q11.2 deletion syndrome
Molecular Psychiatry (2023)
-
Deep exome sequencing identifies enrichment of deleterious mosaic variants in neurodevelopmental disorder genes and mitochondrial tRNA regions in bipolar disorder
Molecular Psychiatry (2023)
-
Structured tracking of alcohol reinforcement (STAR) for basic and translational alcohol research
Molecular Psychiatry (2023)
