
Abstract
Visceral adipose tissue (VAT)—fat stored around the internal organs—has been suggested as an independent risk factor for cardiovascular and metabolic disease1,2,3, as well as all-cause, cardiovascular-specific and cancer-specific mortality4,5. Yet, the contribution of genetics to VAT, as well as its disease-related effects, are largely unexplored due to the requirement for advanced imaging technologies to accurately measure VAT. Here, we develop sex-stratified, nonlinear prediction models (coefficient of determination = 0.76; typical 95% confidence interval (CI) = 0.74–0.78) for VAT mass using the UK Biobank cohort. We performed a genome-wide association study for predicted VAT mass and identified 102 novel visceral adiposity loci. Predicted VAT mass was associated with increased risk of hypertension, heart attack/angina, type 2 diabetes and hyperlipidemia, and Mendelian randomization analysis showed visceral fat to be a causal risk factor for all four diseases. In particular, a large difference in causal effect between the sexes was found for type 2 diabetes, with an odds ratio of 7.34 (95% CI = 4.48–12.0) in females and an odds ratio of 2.50 (95% CI = 1.98–3.14) in males. Our findings bolster the role of visceral adiposity as a potentially independent risk factor, in particular for type 2 diabetes in Caucasian females. Independent validation in other cohorts is necessary to determine whether the findings can translate to other ethnicities, or outside the UK.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The data on which this study is based (application number 15152) are available for bona fide researchers from the UKBB Resource (http://www.ukbiobank.ac.uk/about-biobank-uk/), on filing an application to the UKBB. The data for VAT^ can be accessed via the UKBB Resource, while the summary statistics of the GWAS are available for download from the GWAS Catalog (https://www.ebi.ac.uk/gwas/). Relevant additional data will be available from the authors on request.
References
Fox, C. S. et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 116, 39–48 (2007).
Vega, G. L. et al. Influence of body fat content and distribution on variation in metabolic risk. J. Clin. Endocrinol. Metab. 91, 4459–4466 (2006).
Tchernof, A. & Despres, J.-P. Pathophysiology of human visceral obesity: an update. Physiol. Rev. 93, 359–404 (2013).
Brown, J. C., Harhay, M. O. & Harhay, M. N. Anthropometrically-predicted visceral adipose tissue and mortality among men and women in the third National Health and Nutrition Examination Survey (NHANES III). Am. J. Hum. Biol. 29, 444–454 (2017).
Katzmarzyk, P. T., Mire, E. & Bouchard, C. Abdominal obesity and mortality: the Pennington Center longitudinal study. Nutr. Diabetes 2, e42–e43 (2012).
Bentham, J. et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 390, 2627–2642 (2017).
Samouda, H. et al. VAT = TAAT − SAAT: innovative anthropometric model to predict visceral adipose tissue without resort to CT-scan or DXA. Obesity 21, 41–50 (2013).
Eastwood, S. V. et al. Estimation of CT-derived abdominal visceral and subcutaneous adipose tissue depots from anthropometry in Europeans, South Asians and African Caribbeans. PLoS One 8, e75085 (2013).
Neamat-Allah, J. et al. Validation of anthropometric indices of adiposity against whole-body magnetic resonance imaging—a study within the German European Prospective Investigation into Cancer and Nutrition (EPIC) cohorts. PLoS One 9, e91586 (2014).
Neamat-Allah, J. et al. Can the use of blood-based biomarkers in addition to anthropometric indices substantially improve the prediction of visceral fat volume as measured by magnetic resonance imaging? Eur. J. Nutr. 54, 701–708 (2015).
Fox, C. S. et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 8, e1002695 (2012).
Sung, Y. J. et al. Genome-wide association studies suggest sex-specific loci associated with abdominal and visceral fat. Int. J. Obes. 40, 662–674 (2016).
Chu, A. Y. et al. Multiethnic genome-wide meta-analysis of ectopic fat depots identifies loci associated with adipocyte development and differentiation. Nat. Genet. 49, 125–130 (2017).
Speed, D. et al. Reevaluation of SNP heritability in complex human traits. Nat. Genet. 49, 986–992 (2017).
Sodini, S. M., Kemper, K. E., Wray, N. R. & Trzaskowski, M. Comparison of genotypic and phenotypic correlations: Cheverud’s conjecture in humans. Genetics 209, 941–948 (2018).
Locke, A. E. A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Moreno-Navarrete, J. M. et al. Heme biosynthetic pathway is functionally linked to adipogenesis via mitochondrial respiratory activity. Obesity 25, 1723–1733 (2017).
Gómez-Hernández, A., Beneit, N., Díaz-Castroverde, S. & Escribano, O. Differential role of adipose tissues in obesity and related metabolic and vascular complications. Int. J. Endocrinol. 2016, 1216783 (2016).
Ahlqvist, E. et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 6, 361–369 (2018).
Emdin, C. A. et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. J. Am. Med. Assoc. 317, 626–634 (2017).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data.Nat. Commun. 9, 224 (2018).
Haycock, P. C. et al. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am. J. Clin. Nutr. 103, 965–978 (2016).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction (Springer, 2001).
R Core Development Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2016).
Walter, K. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Weeks, J. P. plink: An R package for linking mixed-format tests using IRT-based methods. J. Stat. Softw. 35, 1–33 (2010).
Aulchenko, Y. S., Ripke, S., Isaacs, A. & van Duijn, C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007).
MacArthur, J. et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901 (2017).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: atool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions.Nat. Commun. 6, 5890 (2015).
Carithers, L. J. & Moore, H. M. The Genotype-Tissue Expression (GTEx) project. Biopreserv. Biobank. 13, 307–308 (2015).
Ferreira, M. A. et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 49, 1752–1757 (2017).
Durinck, S. et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005).
Bulik-Sullivan, B. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Nagano, H. et al. p53-inducible DPYSL4 associates with mitochondrial supercomplexes and regulates energy metabolism in adipocytes and cancer cells. Proc. Natl Acad. Sci. USA 115, 8370–8375 (2018).
Nauli, S. M. et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129–137 (2003).
Bodle, J. C. et al. Primary cilia: the chemical antenna regulating human adipose-derived stem cell osteogenesis. PLoS One 8, e62554 (2013).
Ahsan, M. et al. The relative contribution of DNA methylation and genetic variants on protein biomarkers for human diseases. PLoS Genet. 13, e1007005 (2017).
Berkson, J. Limitations of the application of fourfold table analysis to hospital data. Biometrics Bull. 2, 47–53 (1946).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Bowden, J., Smith, G. D. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Acknowledgements
We acknowledge all of the participants and staff involved in UKBB for their valuable contribution. This research was conducted using the UKBB Resource under application number 15152, following the restrictions on data availability set up by the UKBB. The computations were performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science under projects b2016021 and sens2017538. The research was funded by the Swedish Society for Medical Research (M.R.-A. and Å.J.), Swedish Research Council (Å.J., 2015-03327), Kjell and Märta Beijers Foundation (Å.J.), Göran Gustafssons Foundation (Å.J.), Marcus Borgström Foundation (Å.J.), Åke Wiberg Foundation (Å.J., M16-0210), Swedish Heart and Lung Foundation (Å.J., 20170484), Swedish Diabetes Foundation (C.W.) and Science for Life Laboratory (Å.J.).
Author information
Authors and Affiliations
Contributions
T.K., M.R.-A. and Å.J. designed the study and performed the data analysis. T.K. developed all of the models, performed the statistical analysis and generated the figures. G.P. and C.W. performed the functional study. T.K., M.R.-A., G.P., J.H., C.W., W.E.E. and Å.J. interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Kate Gao and Brett Benedetti were the primary editors on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended Data
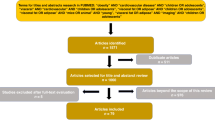
Extended Data Fig. 1 Study selection.
Instances 0, 1 and 2 denote different data collection time periods (instance 0, 2006–2010; instance 1, 2012–2013; instance 2, 2014 to the present). At instance 2, VAT mass was measured by DXA. For the MR analysis, the cohort was split into two nonoverlapping subsets. IV, instrumental variable.
Extended Data Fig. 2 Correlations between predictors and measured VAT mass.
a,b, The length of each bar denotes the strength of the pairwise Pearson’s product–moment correlation between the regression predictor (specified on the left) and measured VAT mass, for the female (a) and male (b) training datasets. The strength of the correlation is also visualized by color (dark blue, lowest correlation; yellow, highest correlation). The regression predictors are ordered from largest positive to largest negative correlation in females. Error bars denote 95% asymptotic CIs based on Fisher’s Z transform. Sample sizes are n = 2,010 for females and n = 2,188 for males.
Extended Data Fig. 3 Bias and 95% CIs of full prediction models, as a function of measured VAT.
a,b, VAT^, as predicted from the leave-one-out cross-validation, plotted against measured VAT mass for females (a; black circles) and males (b; red circles). Also plotted is VAT^ against measured VAT mass for the out-of-sample data (green dots in a and blue dots in b). The out-of-sample datasets constitute Irish and other white individuals from the UKBB, excluding white British. The sample sizes are n = 2,010 females and n = 2,188 males for the training datasets, and n = 119 females and n = 102 males for the out-of-sample datasets. The long-dashed, gray lines denote the linear fits (ordinary least squares regression) to the leave-one-out cross-validation data, and the gray (a) and red shaded areas (b) denote the corresponding CIs of the estimated slopes. Green (a) and blue shaded areas (b) denote the CIs of the linear fits (not plotted) to the out-of-sample data. Thin black lines denote the one-to-one relation. A slope below the one-to-one relation indicates that a small bias is present in the data. However, note that the attenuation is exaggerated due to measurement errors also in the measured VAT mass. c,d, VAT prediction residuals plotted against measured VAT mass for females (c) and males (d). The long-dashed lines correspond to the fitted regression lines in a and b. The gray, solid lines denote the conditional 95% CIs. These lines become dashed at high VAT mass, to indicate an increasing uncertainty in the CIs. Otherwise, symbols are as in a and b.
Extended Data Fig. 4 Bias and 95% CIs of reduced prediction models, as a function of measured VAT.
Symbols and sample sizes as in Extended Data Fig. 3.
Extended Data Fig. 5 Overview of the genomic locations of the selected SNPs in the functional study.
The locations of the SNPs are indicated by vertical lines. a, The HMBS/VPS11 region. Region 1 contains two SNPs (rs2509121 and rs11217133). Region 2 contains five SNPs (rs1784461, rs1786141, rs1784460, rs1784459 and rs1786684). Region 3 contains two SNPs (rs1799993 and rs1006195). b, The PKD1 region. Region 1 contains one SNP (rs13337177). Region 2 contains one SNP (rs36232). c, The DPYSL4 region. Region 1 contains three SNPs (rs881347, rs61865793 and rs11146233).
Extended Data Fig. 6 Results from the luciferase assay in HepG2 cells.
For each set of alleles, the box plots represent the median, interquartile range, and minimum and maximum values of all replicates, except for outliers, which are represented as individual points. The total number of replicates of each plasmid is given by the number of independent plasmid extractions multiplied by the number of independent transfections. The P values (two-sided t-test) represent pairwise differences in means, either for the control plasmid (pGL4.10 or pGL4.23) without any insert versus the same plasmid with one of the fragments inserted, or for the two plasmids with fragments, with different alleles inserted. In the names, the subscripts _E and _P indicate whether the fragment was cloned as an enhancer (_E) or promoter (_P) element, with _P1 and _P2 representing two different fragments in the same promoter region. The last part of the names represents the alleles of the SNPs that were targeted by each fragment (see Supplementary Table 14). Two to three independent plasmid extractions and transfections were performed, with each transfection being replicated three times.
Extended Data Fig. 7 Effect of VAT^ on the risk of developing type 2 diabetes for subgroups with specified medical complications.
a,b, Estimated ORs for females (a) and males (b). The solid, gray lines and 95% CIs (dashed gray lines) correspond to the OR estimated for all female (a) or male cases (b) (see bold text to the left; see also Fig. 2). The black (a) and red vertical lines (b) (with error bars denoting 95% CIs) denote the ORs for the various subgroups with specific medical complications. Note the difference in scale between the two panels. All ORs refer to an increase of 1 kg in VAT^.
Extended Data Fig. 8 Polynomial logistic regression models for heart attack/angina and hyperlipidemia.
a–f, Models for heart attack/angina (a–c) and hyperlipidemia (d–f), showing the probability of disease for the raw, unadjusted data (a and d), the predicted probability of disease for each individual (b and e), given the adopted polynomial model (see Supplementary Table 17), and 95% (basic bootstrap) confidence bands (shaded areas) of the ORs per one-unit increase (1 kg) in VAT^ (c and f), each as a function of VAT^. In a and d, error bars indicate 95% CIs, based on the Poisson statistics. The total numbers of cases and controls for heart attack/angina and hyperlipidemia are given in Fig. 2 for females and males separately. In all panels, black and gray denote females while red and pink denote males.
Extended Data Fig. 9 Sensitivity test for the polynomial logistic regression models.
Each panel shows the log[OR] for five different models that are polynomial in VAT^ to different degrees: dashed lines denote second-degree polynomials (q = 2); dotted lines denote third-degree polynomials (q = 3); long-dashed lines denote fourth-degree polynomials (q = 4); dot-dashed lines denote fifth-degree polynomials (q = 5); and two-dashed lines denote sixth-degree polynomials (q = 6). The lines are also color-coded from light gray (q = 2) to black (q = 6). All models also include age, smoking behavior and 15 principal components as covariates. The models are polynomial in age, with degrees of the polynomials as indicated in Supplementary Table 17. Shaded areas indicate regions of large model uncertainty. Note that for each disease, all models show a very similar functional form of the log[OR], independent of the degree of the polynomial.
Extended Data Fig. 10 Relationship between the effects of the genetic instruments on VAT^ and their effects on disease.
Each panel shows the relationship between the effects on VAT^ and the effects on disease of the n = 44 nearly independent genetic instruments (Supplementary Table 21). Error bars denote the 95% CI (normal approximation) of each effect estimate. Females are denoted in black, while males are denoted in red. Pleiotropic outliers identified by the GSMR analysis that were removed before estimation of the causal effects are shown in gray (Supplementary Table 22). An observed slope is indicative of a causal relationship between VAT^ and disease that is unbiased by confounding. The dashed lines denote the estimated log[OR] values by the gsmr package (Table 1). Note the different scales of the y axes.
Supplementary information
Supplementary Information
Supplementary Text and Supplementary Figs. 1 and 2.
Supplementary Tables
Supplementary Tables 1–22.
Rights and permissions
About this article

Cite this article
Karlsson, T., Rask-Andersen, M., Pan, G. et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med 25, 1390–1395 (2019). https://doi.org/10.1038/s41591-019-0563-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-019-0563-7
This article is cited by
-
Dietary Insulin Index (DII) and Dietary Insulin load (DIL) and Caveolin gene variant interaction on cardiometabolic risk factors among overweight and obese women: a cross-sectional study
European Journal of Medical Research (2024)
-
Defining type 2 diabetes polygenic risk scores through colocalization and network-based clustering of metabolic trait genetic associations
Genome Medicine (2024)
-
Association of metabolically unhealthy non-obese and metabolically healthy obese individuals with arterial stiffness and 10-year cardiovascular disease risk: a cross-sectional study in Chinese adults
Nutrition Journal (2023)
-
BMI-adjusted adipose tissue volumes exhibit depot-specific and divergent associations with cardiometabolic diseases
Nature Communications (2023)
-
Illuminating the ‘healthy obese’ phenotype
Nature Metabolism (2023)
