
Abstract
Most studies of adaptive immunity to SARS-CoV-2 infection focus on peripheral blood, which may not fully reflect immune responses at the site of infection. Using samples from 110 children undergoing tonsillectomy and adenoidectomy during the COVID-19 pandemic, we identified 24 samples with evidence of previous SARS-CoV-2 infection, including neutralizing antibodies in serum and SARS-CoV-2-specific germinal center and memory B cells in the tonsils and adenoids. Single-cell B cell receptor (BCR) sequencing indicated virus-specific BCRs were class-switched and somatically hypermutated, with overlapping clones in the two tissues. Expanded T cell clonotypes were found in tonsils, adenoids and blood post-COVID-19, some with CDR3 sequences identical to previously reported SARS-CoV-2-reactive T cell receptors (TCRs). Pharyngeal tissues from COVID-19-convalescent children showed persistent expansion of germinal center and antiviral lymphocyte populations associated with interferon (IFN)-γ-type responses, particularly in the adenoids, and viral RNA in both tissues. Our results provide evidence for persistent tissue-specific immunity to SARS-CoV-2 in the upper respiratory tract of children after infection.
Similar content being viewed by others


Main
SARS-CoV-2 induces humoral and cellular immune responses in children, primarily noted by assessing antibody and T cell responses in peripheral blood1,2; however, little is known about immune responses to the virus in lymphoid tissues of the upper respiratory tract, where initial infection and viral replication take place3,4. The palatine tonsils and adenoids are secondary lymphoid structures at the mucosal surface of the naso- and oropharynx, in which tissue-specific T and B cell responses to antigens in the upper respiratory tract can be generated5,6. Here, collaborative interactions between follicular helper T (TFH) cells and B cells enable immunoglobulin (Ig) gene class switching and formation of germinal centers (GCs), where B cells undergo somatic hypermutation of Ig genes and affinity maturation and results in the production of high-affinity antibodies and memory B cells. As tonsillectomy and adenoidectomy are among the most common pediatric surgeries, these tissues offer an accessible secondary lymphoid tissue to study immune responses to SARS-CoV-2 (ref. 7). Using in-depth immune profiling, we characterized adaptive immune responses to SARS-CoV-2 in the tonsils and adenoids of children after COVID-19 infection and described antigen-specific responses, as well as long-term alterations in tissue-specific B and T lymphocyte populations involved in GC and antiviral memory responses following COVID-19 infection.
Results
SARS-CoV-2 induces robust GC responses
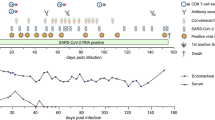
We collected blood, tonsils and adenoids from 110 children who underwent tonsillectomy and/or adenoidectomy primarily between September 2020 and January 2021 in the Washington DC metropolitan area (Fig. 1a and Supplementary Tables 1–3). All participants were required to have a negative SARS-CoV-2 polymerase chain reaction (PCR) test from a nasopharyngeal swab within 72 h before surgery. Eleven participants had a previous diagnosis of COVID-19 confirmed by PCR or antigen detection, ranging from 25 to 303 d before surgery (average 102 d), with only 7 of these 11 participants (64%) reporting symptoms at the time of positive testing (Fig. 1b and Supplementary Table 3). Thirteen additional participants were identified as having been infected with SARS-CoV-2 through serological testing and/or identification of B cells that bound probes for both the S1 and receptor-binding domains (RBD) of the SARS-CoV-2 spike protein (S1+RBD+ B cells), resulting in a total of 24 participants with evidence of previous COVID-19 (post-COV; Fig. 1a and Supplementary Table 4). The remaining 86 participants were used as uninfected controls (UCs).

a, Enrollment of post-COV and UC individuals and study design. b, Time from previous positive SARS-CoV-2 PCR/antigen test to tonsillectomy and/or adenoidectomy in 11 individuals with known previous infection. c, Neutralizing antibody titers (PsVNA50) against the early isolate WA-1 and seven other SARS-CoV-2 variants of interest (post-COV, n = 23; UC, n = 14). d, Correlation between neutralizing antibody titers to WA-1 and days from positive SARS-CoV-2 test to surgery (n = 10). Spearman’s rank correlation (r) and P values are noted. e, Frequency of SARS-CoV-2-specific (S1+RBD+) cells among total CD19+ B cells from PBMCs, adenoids and tonsils from post-COV and UC donors (PBMCs post-COV, n = 18; UC, n = 33; adenoids post-COV, n = 16; UC, n = 27; and tonsil post-COV, n = 16; UC, n = 30; all P < 10−6). f, Representative flow cytometry plots showing the percentage of S1+RBD+ cells among IgD−CD38−CD27+CD19+ switched memory B cells (CD27+ BSM) in post-COV PBMCs, adenoids and tonsils. g, Composition of S1+RBD+ B cells and total B cells from post-COV PBMCs (n = 18), adenoids (n = 16) and tonsils (n = 16). Mean frequency of each B cell subset (defined in Supplementary Figs. 1 and 2) shown in pie chart. ASC, antibody secreting cells equivalent to plasma cells and plasmablasts; CD27+ BUM, CD27+IgD+ unswitched memory B. h, Images of adenoids and tonsils showing GCs from one post-COV donor and one UC, representative of three post-COV and three UC donors. Inset shows close-up of GC and light (CD21, follicular dendritic cells, cyan) and dark zones (Ki-67, dividing cells, red). CD138 (plasma cell and epithelial cell marker) in blue. Scale bars, 1 mm (top), 2 mm (middle) and 200 μm (bottom). i, Composition of S1+RBD+ IgD−CD27−CD38−CD19+ DN B cells and total DN B cells from post-COV PBMCs (n = 18), adenoids (n = 16) and tonsils (n = 16). Mean frequency of each DN subset is shown in bar chart. Mean ± s.d. are displayed in bar and scatter-plots. Each dot represents one donor. Significance calculated with two-sided Mann–Whitney U-test. ****P < 0.0001.
During the sample collection period, the dominant circulating SARS-CoV-2 strains in the Washington DC area were the D614G variant (similar to WA-1) until November 2020 and Alpha after December 2020 (refs. 8,9). Accordingly, neutralizing antibodies against WA-1, B.1.1.7 (Alpha) and B.1.429 (Epsilon) were detected in the serum of all seropositive individuals, but not UCs; fewer post-COV individuals had neutralizing antibodies to other variants of concern, including B.1.617.2 (Delta, 21 of 23) and B.1.1.529 (Omicron, 9 out of 23) (Fig. 1c and Supplementary Table 4). Neutralizing titers were highest against the WA-1 strain and inversely correlated with the time since a positive PCR/antigen test in participants with previous testing (Fig. 1d). Overall, 80% of participants were seropositive to the common cold coronaviruses HCoV-OC43 and HCoV-HKU1, with no differences between post-COV and UC groups (Supplementary Table 4).
We detected S1+RBD+ B cells in peripheral blood mononuclear cells (PBMCs), tonsils and adenoids of all seropositive individuals (Fig. 1e), although responses were heterogeneous. Donors CNMC91 and CNMC104 had very few S1+RBD+ binding B cells in PBMCs and the lowest serum-neutralizing antibody titers to WA-1 among our cohort, whereas another participant (CNMC32) had high serum-neutralizing titers, but very low percentages of S1+RBD+ B cells, particularly in the tonsils and adenoids (Extended Data Fig. 1a).
High-dimensional flow cytometry analyses of B cells from post-COV PBMCs, tonsils and adenoids indicated that the majority of S1+RBD+ B cells were Ig class-switched IgD−CD38−CD27+ memory B cells (hereafter, CD27+ BSM cells) (Fig. 1f,g, Extended Data Fig. 1b and Supplementary Figs. 1 and 2) that were present up to 10 months post-infection (Extended Data Fig. 1c) and were primarily IgG+ (81% in PBMC, 82% adenoids, 84% tonsil; Extended Data Fig. 1d); fewer were IgA+ relative to the total CD27+ BSM cells even in the adenoid and tonsil (14% versus 34% in adenoid, P < 0.001; 14% versus 28% in tonsil, P < 0.001) (Extended Data Fig. 1d). Of note, the percentage of S1+RBD+ B cells (0.2% in adenoids and tonsils) among CD27+ BSM cells in the pharyngeal tissues of post-COV individuals was comparable to that reported in the lung and lung-draining lymph nodes from organ donors who were post-COVID-19 (Extended Data Fig. 1e)10.
Postmortem analyses of adults with fatal COVID-19 revealed loss of GCs in lymphoid organs11; however, we observed similar numbers and sizes of GCs, with discrete dark and light zones, in adenoids and tonsils from post-COV and UC children using multiplex immunofluorescence microscopy (Fig. 1h and Extended Data Fig. 1f,g). We also found a substantial portion of IgD−CD38intCD19+ GC B cells (hereafter, GC B cells) among the S1+RBD+ B cells in both tissues (19% adenoid and 18% tonsil, range 2–47%; Fig. 1g). Paired analyses indicated more S1+RBD+ B cells among both total and GC B cells in adenoids compared to tonsils from the same donor (Extended Data Fig. 1h,i). The frequencies of S1+RBD+ B cells in adenoids, but not tonsils or PBMCs, also correlated significantly with serum neutralization titers for B.1.351 (Beta), B.1.526 (Iota), B.1.617.2 (Delta) and B.1.1.529 (Omicron) variants (Extended Data Fig. 1j), suggesting an important role for the adenoids in generating immune responses to SARS-CoV-2.
Early responses to SARS-CoV-2 in symptomatic patients are dominated by extrafollicular responses, characterized by expansion of IgD−CD27−CD38−CD19+ B cells (double-negative (DN) B cells)12,13. Although we saw expansion of DN B cells among S1+RBD+ B cells in post-COV adenoids and tonsils (Fig. 1g), most were CD21+CD11c− DN B cells (known as DN1 cells; 81% in adenoid and 87% tonsil), which are derived from GCs (Fig. 1i). Only a small proportion (1.3% in adenoid and 1.5% tonsil) were CD21−CD11c+ DN2 B cells, which originate from extrafollicular B cell activation and were reported to expand in acute severe COVID-19 (ref. 12). Thus, robust humoral responses to SARS-CoV-2 are generated and maintained in the tonsils and adenoids after COVID-19 infection.
CITE-seq of S1+ B cells revealed their distinct features
To further investigate B cell responses, we sorted S1-binding (S1+) and non-binding (S1−) B cells from tonsils, adenoids and PBMCs from two post-COV individuals (CNMC71 and CNMC89) and one UC (CNMC99) (Supplementary Fig. 3a,b) and characterized these by cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), which simultaneously measured the expression of 22 B cell surface markers and sequenced the transcriptome and V(D)J/BCR in single cells. Over 1,860 S1+ B cells and 25,000 S1− B cells were captured and analyzed. Surface antibody staining patterns evaluated with unsupervised clustering were concordant with the cell types suggested by gene expression signatures (memory B cells, GC B cells and plasma cells/plasmablasts)14 in each cluster (Fig. 2a–e and Extended Data Fig. 2a,b). Confirming our flow analyses, the majority of S1+ B cells in tonsils and adenoids were in cluster 2, which represented CD27+ BSM cells (Fig. 2c–e) with a smaller, but clear proportion of S1+ B cells in cluster 4, which had a GC B cell gene expression signature and surface protein profile (Fig. 2a–e and Extended Data Fig. 2a,b). In contrast, S1+ B cells in the blood were primarily in cluster 9 (Fig. 2a–c,e), which was a CD27+IgD− cell population (Fig. 2e) but had distinct surface marker and gene expression profile compared to the CD27+IgD−BSM cells in the lymphoid tissues (Fig. 2e and Extended Data Fig. 2a-b). S1+ memory B cells in cluster 2 had higher expression of CXCR3 and HOPX, genes known to be induced by T-bet in T cells15, and lower expression of several inhibitory receptors, including FCGR2B, FCRL2, FCRL3 and TNFRSF13B (encoding TACI)16 than S1− B cells (Supplementary Table 5 and Extended Data Fig. 2c), suggesting distinct features of the SARS-CoV-2-specific B cells.

a, Uniform Manifold Approximation and Projection (UMAP) showing 15 clusters of sorted S1+ and S1− B cells (Supplementary Fig. 3) from tonsils, adenoids and PBMCs from two post-COV (CNMC71 and CNMC89) and one UC (CNMC99) donors clustered according to CITE-seq surface antibody expression. b, Tissue distribution of S1+ and S1− B cells in a. c, Distribution of S1+ B cells among clusters in a. d, Proportion of each of the 15 clusters among S1− and S1+ B cells in a. e, Heat map showing expression of signature gene sets for GC B cells, memory B cells (Mem) and plasma cells/plasmablasts (PC/PB)14 among S1+ B cells organized by cluster. IgD, CD38 and CD27 CITE-seq antibody expression are shown in the bottom heat map in gray. Tissue origin is shown in purple (tonsil), yellow (adenoid) and red (PBMC). Clones shared between tonsil and adenoid are marked in black in the top bar.
BCR sequence analysis confirmed that S1+ B cells were primarily IgG1 and IgA1 class-switched cells (Fig. 3a and Extended Data Fig. 2d), with high frequencies of somatic hypermutation (SHM) in VH genes (Fig. 3b and Extended Data Fig. 2e) and low clonal diversity compared to S1− B cells (Fig. 3c), indicative of antigen-driven clonal expansion and GC origin. Overall, 44 S1+ B cells had the same V and J genes and 80% similarity of their heavy chain CDR3 amino acid (aa) sequence to sequences publicly reported in the CoV-AbDab database17, including one clone similar to 37 published antibody sequences (Supplementary Table 6 and Supplementary Fig. 4a–c).

a, Sub-isotype percentages among sorted S1+ and S1− B cells from PBMC, adenoid and tonsil of one post-COV donor (CNMC89). Labels show the raw number of cells with a given sub-isotype and are only included for sub-isotypes that make up at least 10% of a given category. b, SHM frequency among sorted S1+ and S1− B cells from PBMC, adenoid and tonsil of CNMC89 (PBMC S1+ n = 44, S1− n = 1,491 cells; adenoid S1+ n = 261, S1− n = 1,647 cells; tonsil S1+ n = 416, S1− n = 2,644 cells). Mutation frequency calculated in V gene. Medians ± quartiles and P values are shown in the box plots. c, Simpson’s diversity of S1+ and S1− B cells from PBMCs, adenoids and tonsils from two post-COV donors (CNMC71 and 89) and S1− B cells from one UC (CNMC99). Lower Simpson’s diversity values indicate a greater frequency of large clones. d, Overlap of B cell clones among PBMCs, tonsils and adenoids from post-COV and UC donors. Off-diagonal elements are colored by the Jaccard index of clonal overlap between the two tissues and are labeled by the raw number of overlapping clones. Diagonal elements are labeled by the total number of clones within a particular tissue. e, Clonal lineage trees from two of the largest S1+ B cell clones shared between tonsil and adenoid from CNMC89. Triangles indicate S1+ cells and tip color indicates tissue of origin (purple, tonsil; yellow, adenoid). Isotype and CITE-seq cluster of each cell are listed next to the symbol. Branch lengths represent SHM frequency/codon in VDJ sequence according to the scale bar. Significance calculated with two-sided Mann–Whitney U-test.
A proportion of S1+ B cell clones (83 B cells from 29 clones, 20 clones from CNMC89 and 9 from CNMC71) were present in both the tonsils and adenoids (Fig. 3d). The shared S1+ B cell clones were nearly all isotype-switched (Extended Data Fig. 2f) and consisted primarily of CD27+ BSM cells (cluster 2; Fig. 2e); however, four cells among the shared clones in the tonsil of one donor were GC B cells (cluster 4) (Fig. 2e and Supplementary Table 7). Clonal lineage trees (Fig. 3e) suggested that class switching could occur before, during or after SHM. Thus, multimodal single cell analysis of SARS-CoV-2-specific B cells confirms their emergence from GCs and suggests migration of clonally expanded B cells between pharyngeal lymphoid tissues.
GC populations are expanded post-COVID-19
To determine whether SARS-CoV-2 infection could alter the immune landscape of mucosal tissues beyond acute infection, we used both unsupervised analyses (controlled for age and sex) and manual gating of high-dimensional flow cytometry data to compare immune cell profiles of tonsils, adenoids and PBMCs from post-COV and UC participants (Supplementary Table 2). CD19+ B, CD4+ T and CD8+ T lymphocytes were gated and analyzed independently. Adenoids and tonsils were evaluated together, whereas PBMCs were examined separately, to increase sensitivity for detecting distinct populations in tissues and peripheral blood. Unsupervised analysis of B cells in post-COV versus UC samples revealed more significant differences in cluster frequencies in adenoids than tonsils (Fig. 4a,b and Extended Data Fig. 3a,b). Clusters 3 and 10, representing IgG+ and IgM+ GC B cells, respectively, were significantly increased in post-COV adenoids (Fig. 4b). In addition, a naive-type B cell cluster (cluster 14) was decreased in both post-COV tissues compared to UC (Fig. 4a,b). CD127+IgD+ B cells were also decreased in post-COV PBMCs (Fig. 4c,d and Supplementary Fig. 5a,b), as confirmed by manual gating of CD127+ B cells (Fig. 4e). Thus, changes in B cell populations, including persistent enrichment of GC B cells in the adenoids, were detected after COVID-19 infection.

a–d, Unsupervised clustering of CD19+ B cells from adenoids and tonsils (a) and PBMCs (c) according to flow cytometric surface markers. Quantification of the effect of previous SARS-CoV-2 infection on CD19+ B cell clusters in adenoids and tonsils (b) and PBMCs (d) showing regression coefficients ± 95% confidence intervals (CI) and P values, estimated with a linear model controlling for age and sex. Significantly different clusters (P < 0.05) between post-COV and UC groups are indicated with a star or highlighted in red. Adenoids, post-COV n = 11; UC n = 33; tonsils, post-COV n = 15; UC n = 42; PBMC, post-COV n = 14; UC n = 36. e, Frequency of CD127+ B cells in post-COV (n = 16) and UC (n = 41) PBMCs, P = 0.006. Significance calculated with two-sided Mann–Whitney U-test. Each symbol represents one donor. Mean ± s.d. are displayed. **P < 0.01.
TFH cell populations are expanded after COVID-19
Acute SARS-CoV-2 infection is associated with peripheral T cell lymphopenia18. We found post-COV adenoids had lower percentages of CD3+ and CD4+ T cells compared to UC (29.2% versus 34.1% for CD3+; 23.2% versus 27.9% for CD4+ T cells) (Extended Data Fig. 4a and gating in Supplementary Fig. 6). Unsupervised analysis of CD4+ T cells showed a reduction in cluster 9, which represented CD45RA+CCR7+CD4+ naive cells, in post-COV tonsils and adenoids compared to UC (Fig. 5a,b and Extended Data Fig. 4b,c); decreased percentages of naive CD4+ T cells were also detected by manual gating (Fig. 5c). Conversely, cluster 3, a CD57+PD-1hiCD4+ T cell subset, was significantly enriched in post-COV adenoids and tonsils (Fig. 5a,b), as confirmed by manual gating in adenoids (Fig. 5d). CD57 is a marker of T cell senescence associated with chronic infection, but is also found on some tonsillar CXCR5+PD-1hi GC TFH cells19,20. Compared to the total CD4+ T cell population in adenoids and tonsils from both post-COV and UC individuals, CD57+PD-1hi CD4+ T cells exhibited higher expression of CXCR5, indicative of a TFH cell phenotype and CD69, characteristic of tissue-resident memory T cells (TRM cells) (Fig. 5e)6. Immunofluorescence microscopy indicated that CD57+PD-1hi CD4+ T cells were located within tonsil and adenoid GCs (Fig. 5f) and their frequency positively correlated with the proportion of GC B cells in these tissues (both post-COV and UC samples analyzed) (Extended Data Fig. 5a,b). Phorbol myristate acetate (PMA) and ionomycin stimulation induced production of interleukin (IL)-21 and IL-10, cytokines that facilitate GC formation and B cell antibody secretion, in CD57+PD-1hi CD4+ T cells (Extended Data Fig. 5c,d). Moreover, percentages of CD4+ T cells in cluster 3 positively correlated with percentages of S1+RBD+ B cells that were GC B cells specifically in adenoids (Extended Data Fig. 5e), supporting their role in the generation and persistence of SARS-CoV-2-specific GC responses.

a,b, Unsupervised clustering of CD4+ T cells from adenoids and tonsils according to flow cytometric surface markers (a). Quantification of the effect of previous SARS-CoV-2 infection on CD4+ T cell clusters showing regression coefficients ± 95% CI and P values, estimated with a linear model controlling for age and sex (b). Significantly different clusters (P < 0.05) between post-COV and UC groups are indicated with a star or highlighted in red. Adenoids, post-COV n = 12, UC n = 38; tonsils, post-COV n = 15, UC n = 43. c,d, Frequencies of manually-gated CD45RA+CCR7+ naive CD4+ T cells (P = 0.022 for tonsils) (c) and CD57+PD-1hi CD4+ T cells (P = 0.001 for adenoid) (d) in post-COV and UC adenoids and tonsils (adenoids, post-COV n = 17, UC n = 42; tonsils, post-COV n = 18, UC n = 46). e, Plots of CD69 and CXCR5 expression on CD57+PD-1hi CD4+ T cells and total CD4+ T cells from one tonsil, representative of tonsils and adenoids from 26 donors. f, Image of post-COV adenoid showing CD57+PD-1hi CD4+ T cells in one GC, representative of tonsils and adenoids from six donors. Magnification of square inset shown on the right. CD4 in cyan, CD57 in yellow and PD-1 in magenta. GC boundaries defined using Ki-67 (Fig. 1h). 1 indicates CD4+CD57+; 2 indicates CD4+PD-1+; 3 indicates CD4+CD57+PD-1+ cells. Scale bars, 100 μm (left) and 10 μm (right). g, Cytokine combinations (IFN-γ, IL-2, IL-10, IL-17A, IL-21 and TNF as analyzed by SPICE) produced by tonsillar or adenoid CD4+ T cells from post-COV (n = 13) and UC (n = 13) donors following PMA and ionomycin stimulation (category 27: P = 0.04, 33: P = 0.01, 41: P = 0.03). h,i, Unsupervised clustering of CD4+ T cells from PBMC (h) and quantification of the effect of previous SARS-CoV-2 infection (i) as described in a,b (post-COV n = 13, UC n = 34). j. Frequencies of CD45RA−CXCR5+PD-1+ circulating TFH (cTFH) and CXCR3+CCR6− cTFH cells in post-COV (n = 16) and UC (n = 41) PBMCs, P = 0.032 for CXCR3+CCR6− cTFH cells. Sample list for a–d and h–j in Supplementary Table 2 and for g in Supplementary Table 11. Each symbol represents one donor. Mean ± s.d. displayed in bar plots. Significance calculated with two-sided Mann–Whitney U-test. *P < 0.05, **P < 0.01.
Manual gating also revealed more CD25+CXCR5+PD-1hi cells among CD4+ T cells in post-COV tonsils compared to UC (Extended Data Fig. 5f); these CD25+CXCR5+PD-1hi cells produced more IL-21 and IL-10 after PMA and ionomycin stimulation compared to their CD25− counterparts (Extended Data Fig. 5g) and their frequencies correlated with percentages of GC B cells in the tonsils (Extended Data Fig. 5h), suggesting they were activated cells important for GC generation21. Cluster 6, which contained a population of CD45RA−CXCR5+PD-1int pre-TFH cells that were CXCR3+CCR6− (Extended Data Fig. 4b,c), a combination of markers associated with IFN-γ and TH1 cytokine production22, was also significantly increased in post-COV adenoids (but not tonsils) compared to UC; this was confirmed by manual gating (Fig. 5a,b and Extended Data Fig. 5i). A high percentage of CXCR3+CCR6− pre-TFH cells produced IFN-γ after PMA and ionomycin stimulation (Extended Data Fig. 5j,k), suggesting that type 1 T cell responses were induced as part of the antiviral response to SARS-CoV-2 in adenoids.
Stimulation with PMA and ionomycin revealed several combinations of cytokines had higher expression in CD4+ T cells in post-COV tonsils and adenoids than UC (Fig. 5g and Supplementary Fig. 7). Two of these combinations (categories 33 and 41) included IL-21, suggesting they were produced by TFH cells; category 33 also included IL-10 (Fig. 5g). Increased IFN-γ was part of a cytokine pattern (category 27) specifically enriched in post-COV adenoids compared to UC (Fig. 5g), consistent with the enrichment of CXCR3+CCR6− pre-TFH cells in post-COV adenoids. We also noted more robust IFN-γ production by CD4+ T cells in adenoids than tonsils (post-COV and UC analyzed together) (Extended Data Fig. 5l), indicating inherent differences between T cells in these tissues.
In PBMCs, unsupervised analysis revealed that two clusters (5 and 11) of CD45RA−CXCR5+PD-1+ circulating TFH-like cells (cTFH cells) expressing the activation marker CD38 (ref. 23) were increased in post-COV samples compared to UC (Fig. 5h,i and Extended Data Fig. 6a,b); cluster 11 was also CXCR3+. Although percentages of total cTFH cells were not increased in post-COV PBMCs compared to UC, manual gating indicated that cTFH cells were skewed to a CXCR3+CCR6−phenotype (Fig. 5j and gating in Supplementary Fig. 8); these cells produced IFN-γ upon stimulation with PMA and ionomycin (Extended Data Fig. 6c). We also observed a higher frequency of CD45RA+CCR7+CD28+CD27+CD95+ stem cell-like memory CD4+ T cells (TSCM cells) (Extended Data Fig. 6d) in post-COV PBMCs than UC, perhaps reflecting long-lived memory T cells previously reported in children after COVID-19 infection24. Thus, expansion of functional TFH cells with IFN-γ-associated skewing suggests prolonged immune activation in the upper respiratory tract and PBMCs of children weeks to months after COVID-19 infection.
CD8+ TRM cells are expanded post-COVID-19
Similar to CD4+ T cells, we observed reduced percentages of a naive CD8+ T cell cluster (cluster 1) in adenoids from post-COV donors compared to UC (Fig. 6a,b and Extended Data Figs. 7a,b and 8a). Manual gating revealed a similar, although not significant trend, in both adenoids and tonsils, in addition to more effector memory CD8+ T cells in post-COV tonsils compared to UC (Extended Data Fig. 8b,c). Adenoids and tonsils from post-COV donors exhibited non-statistically significant increases in clusters 2 and 3 compared to UCs, which represented HLA-DR+CD38+CXCR3+CCR7−CD45RA− activated effector memory CD8+ T cells that were either CD38hi or CD57+ (Fig. 6a,b). Manual gating showed significantly more CD57+PD-1+CD8+ T cells in post-COV adenoids and tonsils (Fig. 6c) and more CXCR3+CCR6−CD8+ T cells (Tc1 skewed) in post-COV adenoids than samples from UC (Extended Data Fig. 8d). In addition, CD8+ T cells from adenoids produced more IFN-γ than those from tonsils upon PMA and ionomycin stimulation (post-COV and UC analyzed together) (Extended Data Fig. 8e).

a,b, Unsupervised clustering of CD8+ T cells from adenoids and tonsils according to flow cytometric surface markers (a). Quantification of the effect of previous SARS-CoV-2 infection on CD8+ T cell clusters showing regression coefficients ± 95% CI and P values, estimated with a linear model controlling for age and sex (b). Significantly different clusters (P < 0.05) between post-COV and UC groups are indicated with a star or highlighted in red. Adenoids post-COV n = 12, UC n = 35; tonsils post-COV n = 15, UC n = 42. c, Frequency of CD57+PD-1+CD8+ T cells in post-COV and UC adenoids (post-COV n = 17, UC n = 42, P = 0.044) and tonsils (post-COV n = 18, UC n = 46, P = 0.030) d, Flow cytometry plots showing CD69, CD103, CXCR5 and CXCR3 expression on CD57+PD-1+CD8+ T cells from one tonsil, representative of tonsils and adenoids from 26 donors. e, Adenoid from post-COV donor showing the location of CD57+PD-1+CD8+ T in one GC, representative of six samples. GC is circled, magnification of square is in inset. CD8 is cyan, CD57 is yellow, PD-1 is pink. HLA-DR (blue) stains follicles and Ki-67 (red) stains GC. 1 indicates CD8+CD57+PD-1+; 2 indicates CD8+CD57+ cells. Scale bars, 50 μm (left) and 10 μm (right). f, Cytokine/cytotoxic factor combinations (involving granzyme B, IFN-γ, CD107a, IL-2 and TNF, analyzed by SPICE) produced by tonsillar CD8+ T cells from post-COV (n = 13) and UC (n = 13) donors following PMA and ionomycin stimulation (category 3, P = 0.049; 10, P = 0.051; 14, P = 0.035; 18, P = 0.020; 19, P = 0.032; 22, P = 0.007; 23, P = 0.001; 26, P = 0.017; 30, P = 0.025). Sample list for a–c is in Supplementary Table 2 and e,f in Supplementary Table 11. Each symbol represents one donor. Mean ± s.d. are displayed in bar plots. Significance calculated using two-sided Mann–Whitney U-test. *P < 0.05.
CD8+ T cells expressing CD57 and PD-1 are expanded in the peripheral blood of adults with moderate and severe COVID-19 (ref. 25); however, their function is unclear. CD57+PD-1+CD8+ T cells in post-COV adenoids and tonsils exhibited robust production of IFN-γ, tumor necrosis factor (TNF), IL-2, granzyme B and perforin following PMA and ionomycin stimulation (Extended Data Fig. 8f,g), expressed the tissue-resident markers CD103 and CD69 in addition to CXCR5 (Fig. 6d) and were found in the GC (Fig. 6e). Moreover, tonsillar CD8+ T cells from post-COV donors had higher expression of multiple combinations of cytokines/cytotoxic factors compared to those from UC (Fig. 6f and Supplementary Fig. 9). Thus, activated CD8+ T cell populations were enriched in the pharyngeal lymphoid tissues after COVID-19 infection.
In contrast, the only significant finding among CD8+ T cell populations in PBMCs (Extended Data Fig. 8h,i and Supplementary Fig. 10a,b) were more abundant CD45RA+CCR7+CD28+CD27+CD95+CD8+ TSCM cells seen by manual gating in post-COV relative to UC samples (Extended Data Fig. 8j) as noted by others26. Thus, activated and cytotoxic CD8+ TRM cells in the GC were enriched in the tonsils and adenoids post-COVID-19, whereas fewer significant changes were detected in PBMCs.
Expanded T cell clonotypes may be SARS-CoV-2 specific
To evaluate whether changes in T cell populations reflected SARS-CoV-2 antigen-specific responses, we stimulated post-COV tonsils, adenoids and PBMCs with spike (S), membrane (M) and nucleocapsid (N) peptide pools and assessed activation-induced markers on T cells. SARS-CoV-2-reactive CD4+ T cells in PBMCs had the greatest responses to the S-peptide pool (Fig. 7a,b). Concatenation of all peptide-activated CD4+ T cells indicated that SARS-CoV-2-responsive CD4+ T cells in PBMCs were primarily memory cells that were enriched for CD45RA−CXCR5+PD-1+ cTFH cells that were CXCR3+ (Fig. 7c).

a, Representative flow cytometry plots showing gating of antigen-specific CD4+ T cells from post-COV PBMCs expressing activation-induced markers (AIM+, CD40L+4-1BB+) following stimulation with SARS-CoV-2 peptide pools of S, M and N. Dimethylsulfoxide (DMSO) (vehicle, V) was the negative control, PHA-L was the positive control. b, Frequencies of AIM+CD4+ T cells from six post-COV PBMCs as in a (V versus S, P = 0.031; V versus M, P = 0.031; V versus N, P = 0.031). Significance calculated with two-sided Wilcoxon signed-rank test for paired samples from the same donor. c, Flow cytometry plots showing frequency of memory T cells (shown in box on left), CD45RA−CXCR5+PD-1+ cTFH cells and CXCR3+CCR6− cTFH cells from concatenated antigen-specific CD4+ T cells from S, M and N peptide pool stimulations from six donors compared to total CD4+ T cells in PBMC. d, Frequency of CD8+ T cells that are part of expanded clonotypes (frequency > 0.01, clone defined by identical CDR3β aa sequence) in tonsils, adenoids and PBMCs from two post-COV donors (CNMC71 and CNMC89) and one UC (CNMC99) assessed by CITE-seq and TCR sequencing. e–g, UMAP (e), tissue distribution (f) and CITE-seq surface antibody expression (g) of 16 clusters of CD95+CD8+ T cells from tonsils, adenoids and PBMCs of the three donors in d. h–i, Expanded clonotypes (h) and the distribution of expanded and non-expanded clones across clusters (i) of CD95+CD8+ T cells in e. j, Antigens recognized by four expanded CD8+ T cell clones (each represented by a slice) with CDR3β sequences matching those reported to be SARS-CoV-2-specific in public databases; percentage of cells in each clone noted. Clones recognizing spike epitopes in green and ORF1ab epitopes in red. Clones reported to recognize >1 antigen not shown. Nested epitopes recognized by spike- and ORF1ab-specific TCRs are depicted below the pie chart (Supplementary Table 8). k, Overlap of CD8+ T cell clones among PBMCs, tonsils and adenoids from two post-COV donors and one UC; degree of overlap between TCRα/β CDR3 aa sequences was calculated with the Morisita index (shown in plot), ranging from 0 to 1, with 0 indicating no sharing and 1 indicating full overlap. *P < 0.05.
Due to the highly activated nature of T cells in the adenoids and tonsils even without stimulation, we were unable to precisely identify and phenotype SARS-CoV-2-specific T cells in these tissues using peptide pool stimulations (Extended Data Fig. 9a,b), nor were we able to identify antigen-specific CD8+ T cells in PBMCs, adenoids or tonsils. As an alternative approach, we used single-cell T cell receptor (TCR) sequencing to identify clonally expanded SARS-CoV-2-specific T cells and compared these to TCRβ sequences previously reported to recognize SARS-CoV-2 antigens. Non-naive (activated) CD95+CD8+ T cells and CD95+CD4+ T cells were sorted from tonsils, adenoids and PBMCs from the same two post-COV and one UC individuals described above and characterized by CITE-seq, assessing ten T cell surface markers, the transcriptome and TCR sequences of each cell. Analysis of about 13,000 CD8+ T cells and 12,000 CD4+ T cells indicated a higher frequency of expanded clonotypes (>1% clonotype frequency at the CDR3β aa level) among CD8+ T cells in post-COV tonsils, adenoids and PBMCs compared to UC samples (Fig. 7d). By unsupervised clustering of cell surface protein expression, expanded clonotypes were primarily in five clusters (Fig. 7e–i). Clusters 2, 6 and 9 represented CD57+CD8+ T cells from PBMCs (Fig. 7e–g), corresponding to T cell subsets reported to be enriched in peripheral blood during acute COVID-19 (refs. 25,27). Clusters 1 and 12 contained activated CD38+HLA-DR+ TRM cells expressing CXCR5 and PD-1 from tonsils and adenoids, with cluster 12 additionally expressing CD57 (Fig. 7e–g), resembling the activated CD8+ TRM cells we found enriched in post-COV tonsils and adenoids, suggesting these represented clones that expanded in response to SARS-CoV-2.
We then compared TCR CDR3β aa sequences in the expanded clones to those previously reported in databases of SARS-CoV-2-specific TCR sequences (immuneCODE28 and VDJdb29). In one post-COV participant with symptomatic COVID-19 71 d before surgery (CNMC71), 24% of the expanded CD8+ TCRs matched sequences in these databases (13% tonsil, 10% adenoid, 30% PBMC), including the most abundant clonotype (111 cells, detected in PBMCs) (Supplementary Table 8 and Extended Data Fig. 9c,d). Although we identified TCRβ sequences reported to recognize a wide variety of SARS-CoV-2 epitopes among all activated CD8+ T cells (Extended Data Fig. 9e), among the expanded clones, S and ORF1ab were the primary antigens recognized (Fig. 7j), similar to other studies27,30. The S epitopes recognized by these expanded clonotypes were located in the S2 subunit and were among the most reported immunodominant epitopes recognized by CD8+ T cells (Supplementary Table 8)30.
We observed fewer clones with >1% frequency among CD4+ T cells (Supplementary Table 8), suggesting less proliferation among CD4+ T cells compared to CD8+ T cells. Therefore, we used a less stringent definition for expanded CD4+ T cells clones (frequency >0.1% and absolute count ≥3 in a sample). In addition, because more prominent clonally expanded motifs have been noted among TCRα sequences than among TCRβ in SARS-CoV-2-specific CD4+ T cells31, we analyzed both TCR chains (Extended Data Fig. 9f,g and Supplementary Table 8). Unsupervised clustering of activated CD95+CD4+ T cells showed that expanded clonotypes were primarily in cluster 12 in PBMCs, which represented CD57+PD-1+CD4+ T cells (Extended Data Fig. 9h–l) with higher expression of TH1 and cytotoxic genes, including IFNG, TBX21, CCL4, NKG7, PRF1 and GZMB compared to other PBMC clusters (Supplementary Table 8), similar to other studies25,32,33,34. In tonsils and adenoids, most expanded clones were in the TFH-like clusters 1 and 4 (Extended Data Fig. 9h–l). Differential gene expression indicated that cluster 4 had high IFNG expression (Supplementary Table 8).
Among the expanded CD4+ T cell clones, four had CDR3β aa sequences present in the TCRβ-centric database immuneCODE and recognized epitopes in ORF1ab, ORF7b, ORF10 and S (Supplementary Table 8). We also identified ten clones with SARS-CoV-2-specific TCRα sequences reported in VDJdb and the literature31,32,33,35, including the most abundant clone (14 cells from CNMC89 PBMCs), which was paired with the most highly-expanded CD4+ TCRβ sequence (Extended Data Fig. 9m and Supplementary Table 8). Several of the SARS-CoV-2-specific CDR3α sequences we found were shared among two donors (Supplementary Table 8), suggesting that they may recognize immunodominant or public epitopes.
Furthermore, we found overlap of CD8+ T cell clones in post-COV tonsils and adenoids, including some that were SARS-CoV-2-reactive (Fig. 7k and Supplementary Table 8); less clonotype overlap was noted among CD4+ T cells (Extended Data Fig. 9n), perhaps due to their limited expansion. Thus, SARS-CoV-2-specific T cells in the blood and tissue showed persistent clonal expansion and significant sharing of CD8+ T cell clones among tonsils and adenoids after COVID-19 infection.
SARS-CoV-2 viral RNA persisted in post-COV tissue
Because we observed prolonged immune activation and clonal expansion after COVID-19, we assessed viral RNA persistence in pharyngeal lymphoid tissues. Using droplet digital PCR, we found SARS-CoV-2 nucleocapsid RNA in 7 out of 9 adenoid and 15 out of 22 tonsil formalin-fixed, paraffin-embedded tissue blocks from post-COV individuals, despite negative nasopharyngeal swab PCRs at the time of surgery (Fig. 8a and Supplementary Table 9). Viral RNA was not found in any UC (Fig. 8a). In four post-COV donors, their nasopharyngeal swab PCR had been positive over 100 d before surgery, including one 303 d before surgery. Viral RNA copies significantly correlated with the percentages of S1+RBD+ B cells among GC B cells in post-COV tonsils (Fig. 8b), raising the possibility that antigen persistence contributes to prolonged lymphoid and GC responses in post-COV donors.

a, Quantification of SARS-CoV-2 nucleocapsid RNA by droplet digital PCR (ddPCR) from adenoid and tonsil formalin-fixed paraffin-embedded (FFPE) tissue blocks (adenoids post-COV n = 9 and UC n = 6; tonsils post-COV n = 22 and UC n = 9). N1 and N2 represent two regions of the gene encoding the SARS-CoV-2 nucleocapsid. Each symbol represents one donor. Mean ± s.d. are displayed. Analyzed samples are listed in Supplementary Table 9. b, Summary of correlations among various subsets of SARS-CoV-2 antigen-specific B cells, serum-neutralizing antibody titers and T cell populations of interest versus copies of nucleocapsid (N1 and N2) RNA in post-COV tonsils. Correlations assessed with Spearman’s rank correlation (copies N1 versus N2 P < 10−5; percentage of S1+RBD+ B cells among GC B cells versus N1 P = 0.004 and versus N2 P = 0.005). P values were not corrected for multiple comparisons. **P < 0.01; ***P < 0.001.
Discussion
Using samples from pediatric tonsillectomies and adenectomies, we found evidence of persistent immune responses to SARS-CoV-2 in the pharyngeal lymphoid tissues, including antigen-specific memory B and T cells and prolonged changes in lymphocyte populations after infection. The high percentage of seropositive children in our cohort in late 2020 to early 2021, before vaccine availability, underscored the extent of COVID-19 in this urban population36. The variation in memory B cell frequencies and serum-neutralizing antibody titers we observed further highlighted heterogeneity of responses that may leave some children prone to repeat infection. Whether immunization generates immunity to SARS-CoV-2 in the upper respiratory tract and how this compares to natural infection are important questions.
Lasting changes in immune cell populations in the PBMCs and nasal mucosa of adults have been reported months after COVID-19 (refs. 37,38,39,40). We saw more prominent changes in the pharyngeal tissues compared to PBMCs and many of the enriched lymphocyte populations we noted in the tissues were tissue-resident populations that remain at these sites for months and even years41,42. These populations, including TFH cells and CD8+ TRM cells, some of which were likely SARS-CoV-2-specific, exhibited an IFN-γ-type bias that likely led to upregulation of CXCR3 and HOPX in SARS-CoV-2-specific B cells. Strong local type 1 and type 2 IFN responses have been reported in the airways of infected children, which may lead to enhanced viral control compared to adults43. Many of the expanded populations we noted in the tissues expressed CXCR5 and were located in GCs, including CXCR5+CD8+ T cells, which resemble stem-like progenitor cells that maintain antiviral responses in chronic viral infections44,45,46. We also found enrichment of various CD57+ T cell populations, which are found following repeated antigen exposure in chronic infections47. The role of these CXCR5+ and CD57+ cells in the response to an acute respiratory virus like SARS-CoV-2 is less clear, but their enrichment raises the question of whether prolonged antigen exposure contributes to these expanded populations48.
Longitudinal studies suggest continued affinity maturation of SARS-CoV-2-specific B cells in GCs months after infection, possibly due to antigen persistence13,38,49; however, few studies have demonstrated SARS-CoV-2-specific GCs10,50. Our analyses provide direct evidence of ongoing SARS-CoV-2-specific GC reactions with expanded TFH cell populations in adenoids and tonsils weeks to months after acute infection.
Our evaluation of multiple tissues from the same individual further revealed immunologic connections among the pharyngeal lymphoid tissues that may mediate tissue immunity. These findings parallel previous studies that noted B and CD8+ T cell clones distributed across multiple lymph tissues, whereas CD4+ T cell clones were more restricted in distribution51,52,53,54. Nonetheless, our results indicate more significant changes in adenoids than tonsils following COVID-19 infection. Adenoids are located in the nasopharynx and have a respiratory epithelium, whereas palatine tonsils are located in the oropharynx and have a stratified squamous epithelium. These factors, as well as differences in immune cell populations55, may make adenoids more susceptible to immune activation during respiratory infections such as COVID-19, but also raise questions as to whether adenoidectomy and/or tonsillectomy affect immune responses to SARS-CoV-2.
A limitation of our study is the lack of information about dates of infection and symptoms in participants who were unaware of having COVID-19. We also do not have longitudinal samples to precisely map the duration of immunological changes; instead, we relied on time from positive testing to surgery as a proxy. Although we could not identify antigen-specific T cells in the tonsils and adenoids by peptide stimulation due to T cell activation in these chronically inflamed environments, we identified potential antigen-specific T cells by matching TCR sequences to those publicly previously reported to recognize SARS-CoV-2. Lastly, COVID-19-convalescent participants underwent tonsillectomy for sleep-disordered breathing (SDB) or obstructive sleep apnea (OSA) due to hypertrophy of the adenoids and/or tonsils, which may influence local immune responses to SARS-CoV-2 (ref. 50); we used control samples from children with the same conditions to address this concern.
Our findings offer insights into how viral infections shape the mucosal immune tissues in children; maintenance of activated tissue-resident T cells may aid responses against future infectious insults; however, activated cells in these tissues after infection may also contribute to delayed or prolonged sequelae of COVID-19, including long-COVID-19 and multisystem inflammatory syndrome in children, which is characterized by IFN-γ-induced signatures in PBMCs and has mucocutaneous findings, including pharyngeal erythema56,57. Our repository of pharyngeal tissues may facilitate evaluation of these and other important questions.
Methods
Ethics statement
This study was approved by the Institutional Review Board at Children’s National Hospital (protocol no. 00009806). Written informed consent was obtained from parent/guardians of all enrolled participants and assent was obtained from minor participants over 7 years of age.
Participant recruitment
We recruited 110 children undergoing tonsillectomy and/or adenoidectomy at Children’s National Hospital (CNH). All children scheduled to undergo tonsillectomy at CNH were eligible. The first 102 participants were recruited from late September 2020 to early February 2021 without screening for previous COVID-19. An additional two participants were subsequently recruited with a known history of COVID-19, plus six additional individuals (one of whom turned out to be positive by serology) were recruited in May and June 2021. Because not all tissues or blood were available from each individual, we collected a total of 106 blood samples, 100 adenoids and 108 tonsils from 110 participants (Supplementary Table 2). No statistical methods were used to predetermine sample size. All participants had negative PCR with reverse transcription (RT–PCR) test from a nasopharyngeal swab for SARS-CoV-2 within 72 h of surgery. Demographic information and clinical data were collected through parental questionnaires and chart review and managed in REDCap, and biological samples were acquired in the operating room by the clinical team at CNH.
Eleven participants had previous confirmed SARS-CoV-2 infection with RT–PCR or antigen testing from nasopharyngeal swabs. Another 13 COVID-19-exposed participants were identified through serum antibody testing and/or identification of B cells that recognize the spike protein of SARS-CoV-2 by flow cytometry (described below). One participant (CNMC43) had SARS-CoV-2 detected by RT–PCR from the nasopharynx 20 d before surgery but had negative serology and no SARS-CoV-2-specific B cells in the tissue or blood. We excluded this individual from our subsequent analysis.
Control selection within the cohort
Controls for flow cytometric analyses were selected among individuals with no serological or cellular evidence of previous COVID-19. The primary indication for tonsillectomy in all 24 participants with previous COVID-19 was adenotonsillar hypertrophy leading to SDB or OSA (Supplementary Tables 1 and 3) except one participant who had eustachian tube dysfunction. Patients with SDB and OSA both have breathing difficulties during sleep (primarily snoring); however, patients with OSA had polysomnography documenting an apnea–hypopnea index greater than 1, whereas those with SDB did not undergo polysomnography testing and were diagnosed by clinical history alone. None of the 24 participants with prior COVID-19 had frequent recurrent tonsillitis (more than six episodes in a year) or other medical problems that directly affect the immune system aside from atopic disease, nor did they take immunomodulating medications aside from nasal/inhaled steroid or loratadine within 2 weeks of surgery. Therefore, individuals were excluded from the control group if they (1) had periodic fever, recurrent tonsillitis or chronic tonsillitis as primary indication for surgery (n = 15); (2) had more than six episodes of tonsillitis in a year (n = 2); (3) took immunomodulatory medications (including montelukast and cetirizine) aside from inhaled steroid or loratadine within 2 weeks of surgery (n = 9); (4) had sickle cell anemia (n = 3); or (5) did not have flow cytometry studies performed on their samples on the day of processing due to sample collection before panel finalization or technical problems with the flow cytometer on the day of acquisition. Controls were also excluded if they had indeterminate serological testing for SARS-CoV-2 infection and did not have any SARS-CoV-2-specific B cells in the tissue or blood (n = 2); both of these participants subsequently had negative neutralizing titers to SARS-CoV-2 as well. Samples included in unsupervised and manual gating analyses of flow cytometry data are listed in Supplementary Table 2.
Blood and tissue collection
Blood samples were obtained just before the surgical procedure in the operating room in serum separator tubes (BD) for serum collection and sodium heparin tubes (BD) for PBMC extraction. Once received in the laboratory, serum separator tubes were spun at 1,200g for 10 min and serum was aliquoted and stored at −80 °C. PBMCs were isolated the day after collection by density gradient centrifugation on lymphocyte separation medium (MP Biomedicals) at 300g for 30 min at room temperature with no brake and washed with PBS. If red blood cell contamination was present, cells were lysed with ACK buffer.
Tonsils and adenoids were stored in RPMI medium with 5% FBS (VWR), gentamicin 50 mg ml−1 (Gibco) and 1× antibiotic/antimycotic solution (Gibco) on ice immediately after collection. Tissues were processed the day after collection. A 3–5-mm portion of tonsil and adenoid tissue was cut and fixed in 5 ml of 10% buffered formalin (Avantik) for 24–48 h. The fixed tissue was then incubated in 70% ethanol until it was paraffin embedded. The remainder of the tissue was mechanically disrupted and filtered through a 100-μm cell strainer to create a single cell suspension, lysed with ACK buffer (Gibco) and washed with PBS three times. Freshly isolated PBMCs and tonsil and adenoid cells were surface stained and analyzed with flow cytometry as described below on the day of processing. The remaining cells were stored in liquid nitrogen in the presence of FBS with 10% DMSO.
SARS-CoV-2 serum antibody ELISA
After thawing frozen serum to room temperature, IgG and IgM antibodies against the S protein and RBD of the S protein of SARS-CoV-2 were analyzed using ELISA as previously described58,59. Positivity thresholds were based on mean optical density (absorbance) plus 3 s.d. The final criterion of S+ and RBD+ for any combination of positive IgG or IgM gave estimated sensitivity and specificity of 100% based on previous studies of this assay. Data are shown in Supplementary Table 4.
Pseudovirus neutralization assay
Antibody preparations were evaluated by SARS-CoV-2 pseudovirus neutralization assay (PsVNA) using WA-1, B.1.429 (Epsilon), B.1.1.7 (Alpha), P.1 (Gamma), B.1.351 (Beta), B.1.526 (Iota), B.1.617.2 (Delta) and B.1.1.529 (Omicron) strains. The PsVNA using the 293-ACE2-TMPRSS2 cell line was described previously60,61,62.
Briefly, human codon-optimized complementary DNA encoding SARS-CoV-2 S glycoprotein of the WA-1, B.1.429, B.1.1.7, P.1, B.1.351, B.1.526, B.1.617.2 and B.1.1.529 strains were synthesized by GenScript and cloned into eukaryotic cell expression vector pcDNA 3.1 between the BamHI and XhoI sites. Pseudovirions were produced by co-transfection Lenti‐X 293T cells with psPAX2 (gag/pol), pTrip-luc lentiviral vector and pcDNA 3.1 SARS-CoV-2-spike-DeltaC19, using Lipofectamine 3000. The supernatants were collected 48 h after transfection and filtered through 0.45-µm membranes and titrated using 293T-ACE2-TMPRSS2 cells (HEK 293T cells that express ACE2 and TMPRSS2 proteins).
For the neutralization assay, 50 µl of SARS-CoV-2 S pseudovirions were pre-incubated with an equal volume of medium containing serum at varying dilutions at room temperature (RT) for 1 h, then virus–antibody mixtures were added to 293T-ACE2-TMPRSS2 cells in a 96-well plate. The input virus with all SARS-CoV-2 strains used in the current study were the same (2 × 105 relative light units per 50 µl per well). After a 3-h incubation, the inoculum was replaced with fresh medium. Cells were lysed 24 h later and luciferase activity was measured using luciferin. Controls included cells only, virus without any antibody and positive sera. The cutoff value or the limit of detection for the neutralization assay was 1:10. Data are in Supplementary Table 4.
High-dimensional flow cytometry of SARS-CoV-2-specific B cells
Five million cells per sample of PBMC, adenoid or tonsil were resuspended in PBS with 2% FBS and 2 mM EDTA (FACS buffer). Biotinylated S1 and RBD probes (BioLegend) were crosslinked with fluorochrome-conjugated streptavidin in a molar ratio of 4:1. Fluorochrome-conjugated streptavidin was split into five aliquots and conjugated to biotinylated S1 and RBD probes by mixing for 20 min per aliquot at 4 °C. Cells were first stained with the viability dye, Zombie NIR (1:800 dilution, BioLegend) for 15 min at RT, washed twice and then incubated with True-Stain Monocyte Blocker (BioLegend) for 5 min. An antibody cocktail containing the rest of the surface antibodies, the fluorochrome-conjugated S1 and RBD probes and Brilliant Stain Buffer Plus (BD) were then added directly to the cells and incubated for 30 min at RT in the dark (200 μl staining volume). Cells were washed three times and fixed in 1% paraformaldehyde for 20 min at RT before washing again and collecting on a spectral flow cytometer (Aurora, Cytek) using SpectroFlo software (Cytek v.1.1). Antibodies are listed in Supplementary Table 10.
Broad 37 parameter immunophenotyping flow cytometry panel
Two million cells per sample of PBMCs and 5 million cells per adenoid or tonsil were resuspended in FACS buffer. Cells were first stained with LIVE/DEAD Blue (1:800 dilution, Thermo Fisher) for 15 min at RT, washed twice and then incubated with True-Stain Monocyte Blocker (BioLegend) for 5 min. Antibodies for chemokine receptors and TCRγδ were sequentially added at RT (anti-CCR7 for 10 min, anti-CCR6, anti-CXCR5 and anti-CXCR3 together with Brilliant Stain Buffer Plus for 5 min and anti-TCRγδ for 10 min). An antibody cocktail containing the rest of the surface antibodies and Brilliant Stain Buffer Plus was then added to the cells and incubated for 30 min at RT (total staining volume 182 μl). Cells were washed three times and stained with fluorescence-conjugated streptavidin for 15 min at RT. Then, cells were washed twice and fixed in 1% paraformaldehyde for 20 min at RT before washing again and acquiring on the Aurora spectral cytometer (Cytek) using SpectroFlo software (Cytek v.1.1). Antibodies are listed in Supplementary Table 10. Manual gating for both panels was conducted with FlowJo Software v.10 (BD Biosciences) based on previously described gating strategies63.
Unsupervised analysis and statistical modeling
Data from the broad immunophenotyping flow cytometry panel with 37 parameters were analyzed with unsupervised clustering of surface antibody staining. CD19+ B cells, CD4+ T cells and CD8+ T cells were analyzed separately. Tonsils and adenoids were merged and processed together, whereas PBMCs were processed separately due to pre-determined antibody concentration differences in staining required for optimal results in each organ. B cell analyses were based on surface expression of CCR6, CXCR5, CXCR3, CCR7, CD45RA, CD11c, IgD, CD20, IgM, IgG, CD27, HLA-DR, CD38, CD21, CD123, PD-1, CD57, CD25, CD24, CD95, IgA, CD1c, CD127 and CD161. CD4+ and CD8+ T cells analyses were based on the expression of CCR6, CXCR5, CXCR3, CCR7, CD45RA, CD161, CD28, PD-1, CD57, CD25, CD95, CD27, CD127, HLA-DR, CD38, ICOS, CD11c, CD24, CD1c, CD123 and CD21. FCS files (3.0) as well as FlowJo workspaces (v.10.7.2) were processed in R (v.4.1) via Rstudio (v.1.4.1717) and Bioconductor (v.3.13) using cytoverse (v.0.0.0.9000), including flowCore (v.2.4.0), flowWorkspace (v.4.4.0), ggcyto (v.1.20.0), openCyto (v.2.4.0), CytoML (v.2.4.0), cytolib (v.2.4.0) and cytoqc (v.0.99.2). Default options for biexponential data transformation were used. Outlier cells with expression values in the top or bottom 1 × 10−3 quantiles were excluded. Single cells in each sample were first clustered using k-means (k = 500, referred to as metacells), followed by merging cluster centroids from different samples with the same staining (tonsil/adenoids versus PBMC) for meta-clustering and dimensionality reduction. Specifically, 500 centroids from each sample (metacells) were merged followed by another run of k-means meta-clustering (again k = 500), which were finally used in Leiden clustering and to learn a t-UMAP model to project the metacells (single-cell level k-means centroids; shown in plots). Seurat (v.4.0.3), uwot (v.0.1.10) and Leiden (v.0.3.9) were used in shared nearest neighbors graph building, t-UMAP projection and meta-clustering, respectively, with default settings. Leiden meta-clusters were mapped back to the single-cell level and the ranked frequency of single cells in each Leiden meta-cluster in each sample was modeled linearly as a function of age, sex and history of COVID-19 (COVID status) (as in lm(rank(frequency) ~ age + sex + status). Before statistical modeling, principal-component analysis of frequencies was used to detect and exclude outlier samples. Sample sizes are described in the legend of each plot. t-UMAP projections as well as all CIs of coefficients and their P values (from two-tailed Student’s t-tests of each coefficient within each model) are presented in plots built with ggplot2 (v.3.3.5). Data are in Supplementary Table 12.
Processing for CITE-seq
Banked PBMCs, tonsils and adenoids from two post-COV donors (CNMC71 and CNMC89) and one UC (CNMC99) were thawed from liquid nitrogen in a 37 °C water bath for 2–3 min. A total of 2 ml of medium consisting of RPMI with 10% of fetal bovine serum, 0.1 mg ml−1 DNase I (Roche) and 10 mM HEPES was added drop by drop to the thawed cells. Cells were further diluted by incremental addition of a 1:1 volume of medium up to 8 ml, then centrifuged at 300g for 5 min. Cells were then resuspended in 300 μl of medium, incubated at RT for 5 min, washed with medium without DNase I and filtered through a 100-μm strainer before spinning down and resuspending in staining buffer (PBS + 1% BSA). Cells were then incubated with Fc blocker (Human TruStain FcX, BioLegend), stained with TotalSeq-C human hashtag antibodies (BioLegend) to uniquely label the sample origin (by tissue and donor) and washed with PBS + 0.04% BSA. Adenoids and tonsils from the three donors (six samples in total) were pooled together and PBMCs from three were pooled together separately. The number of cells to pool from each tissue and donor was calculated with the aim of pooling a similar number of S1+ B cells from each sample. Pooled cells were first incubated with Fc blocker at 4 °C for 10 min followed by CITE-seq and sorting antibody cocktails in the following order at 4 °C: TotalSeq anti-CXCR3 antibody for 10 min, TotalSeq chemokine cocktail (anti-CCR7, CCR6, CXCR5 antibodies) for 10 min and the rest of CITE-seq antibodies and fluorescence-labeled sorting antibodies and viability dye (Aqua) for 30 min (Supplementary Table 10). Cells were then washed with PBS + 0.04% BSA and resuspended in PBS + 2% FBS. S1+ and S1− B cells, CD95+CD4+ and CD95+CD8+ T cells were sorted from each pool on a BD FACS Aria Fusion sorter for tonsil/adenoid pool and FACS Aria Ill sorter for the PBMC pool (BD Biosciences). Supplementary Fig. 3 details the sorting strategy. Cells were sorted into PBS + 2% FBS. Note that the antibody concentrations used for CITE-seq were optimized by the manufacturer based on healthy PBMC samples and thus may not be optimal for tissue samples. We have not independently verified the specificity of each antibody in our CITE-seq panel. Antibody concentrations were based on our titration from flow cytometry64,65.
Sorted S1+ and S1− B cells and CD95+CD4+ and CD95+CD8+ T cells were mixed with the reverse transcription mix and partitioned into single cell Gel-Bead in Emulsion (GEM) using 10x 5′ Chromium Single Cell Immune Profiling Next GEM v2 chemistry (10x Genomics). The reverse transcription step was performed in an Applied Materials Veriti 96-well thermocycler. 10x Genomics 5′ single-cell gene expression, cell surface protein and BCR or TCR libraries were prepared as instructed by 10x Genomics user guides (https://www.10xgenomics.com/resources/user-guides/). RNA quality and quantity in the libraries were measured using a bioanalyzer (Agilent) and a Qubit fluorometer (Thermo Fisher). Libraires were pooled at a concentration of 10 nM and sequenced on Illumina NovaSeq platform (Illumina) using the following read lengths: Read 1, 26 bp; Index 1, 10 bp; Index 2, 10 bp; Read 2, 150 bp.
CITE-seq data processing and analysis
CellRanger (10x Genomics) v.6.0.0 was used to map cDNA libraries to the hg19 genome reference (10x Genomics hg19 CellRanger reference, v.1.2.0) and to count antibody tag features. Data were further processed using Seurat (v.4.0.1)66 running in R v.4.0.3. After transforming the surface protein library counts using dsb67, we demultiplexed the pooled samples using manual cutoffs on the hashtag antibody staining. We removed cells with fewer than 100 detected genes, greater than 30% mitochondrial reads or mRNA counts greater than 25,000. To exclude cells with extremely high surface antibody counts, we also removed the top 0.05% of cells in the surface antibody total count distribution. Cell clustering was performed by applying the FindNeighbors() function from Seurat on a distance matrix generated from the dsb-transformed surface protein data, followed by Louvain clustering on the resulting shared nearest neighbor graph using Seurat’s FindClusters() algorithm, with a resolution parameter of 1. Expression of selected genes was visualized using the ComplexHeatmap package68 and the percentage of cells per cluster for the S1+ and S1− cells and T cell populations of interest was plotted using ggplot2 (ref. 69). For the comparison of differentially expressed genes between the S1+ and S1− B cells, we first downsampled the fastq files from the S1+ sequencing library to more closely match the reads per cell obtained in the S1− sequencing libraries using seqtk v.1.3. Differential expression was then compared using the MAST algorithm with ‘Donor’ as a latent variable, as implemented in the Seurat FindMarkers function. For RNA-based clustering S1+ and S1− B cells, we first downsampled the fastq files from the S1+ sequencing library to more closely match the reads per cell obtained in the S1− sequencing libraries using seqtk v.1.3. Cells were then clustered using the top 15 principal components derived from the 2,000 most variable genes, selected by Seurat’s FindVariableFeatures function using the ‘vst’ method. Clustering was performed using the Louvain method and a resolution of 1.15 in Seurat’s FindClusters function.
BCR sequence analysis and clonal clustering
BCR repertoire sequence data were analyzed using the Immcantation (www.immcantation.org) framework. Starting with filtered CellRanger output, V(D)J genes for each sequence were aligned to the IMGT GENE-DB reference database v.3.1.29 (ref. 70) using IgBlast v.1.16.0 (ref. 71) and Change-O v.1.0.0 (ref. 72). Nonproductive sequences, cells without associated constant region calls, cells identified as arising from doublets or negative wells and cells with multiple heavy chains were all removed. Samples within each individual were pooled and sequences were grouped into clonal clusters, which contain B cells that relate to each other by somatic hypermutations from a common V(D)J ancestor. Sequences were first grouped by common IGHV gene annotations, IGHJ gene annotations and junction lengths. Using the hierarchicalClones function of scoper v1.1.0 (ref. 73), sequences within these groups differing by a length normalized Hamming distance of 0.1 within the CDR3 region were defined as clones using single-linkage hierarchical clustering74. This threshold was determined through manual inspection of distance to nearest neighbor plots using shazam v.1.1.0 (ref. 75). These heavy chain-defined clonal clusters were further split if their constituent cells contained light chains that differed by V and J genes. Within each clone, germline sequences were reconstructed with D segment and N/P regions masked (replaced with ‘N’ nucleotides) using the createGermlines function within dowser v.0.1.0 (ref. 76). All BCR analyses used R v.4.1.1 (R Core Team 2017) and plots were generated using ggpubr v.0.4.0 (ref. 77) and ggplot2 v.3.3.5 (ref. 69). After clonal clustering, only heavy chain sequences were used for subsequent analysis. Somatic hypermutation was calculated as the Hamming distance between each sequence’s IMGT-gapped sequence alignment and its predicted unmutated germline ancestor along the V gene (IMGT positions 1–312).
Clonal diversity is an important metric of B cell repertoires and low B cell clonal diversity is consistent with an adaptive immune response. To quantify B cell clonal diversity, we calculated Simpson’s diversity for each sample using the alphaDiversity function of alakazam v.1.1.0 (ref. 72). Lower values of Simpson’s diversity indicate a greater probability of two random sequences belonging to the same clone, consistent with more large clones. To account for differences in sequence depth, samples within each comparison were downsampled to the same number of sequences and the mean of 1,000 such re-sampling repetitions was reported. Only donor/tissue/cell sort samples with at least 100 B cells were included, which led to the exclusion of all S1+ cells from CNMC99 (UC) and S1+ PBMCs from CNMC89 (post-COV). Clonal overlap among tissues can be used as a measure of immunological connectivity. Clonal overlap was calculated using the Jaccard index, which for each pair of tissues is the number of unique clones found in both tissues (intersect) divided by the total number of unique clones among the two tissues (union). Clones were labeled as ‘S1+’ if they contained at least one S1+ sorted B cell. To infer lineage trees, we estimated tree topologies, branch lengths and individual-wide substitution model parameters using maximum likelihood under the GY94 model78,79. Using fixed tree topologies estimated from the GY94 model, we then estimated branch lengths and donor-wide parameter values under the HLP19 model in IgPhyML v.1.1.3 (ref. 78). Trees were visualized using dowser v.0.1.0 and ggtree v.3.0.4 (ref. 80).
To identify convergent BCR sequences, heavy chain sequences were compared to previously published SARS-CoV-2 binding antibodies in the CoV-AbDab database17. BCR sequences were identified as convergent with a previously published antibody if they used the same V gene, J gene, CDR3 length and had an aa Hamming distance of no more than 20% in the CDR3.
TCR sequence analysis
TCR repertoire sequence data were analyzed using the scRepertoire package v.1.5.2 (ref. 81) in R v.4.1.1 (R Core Team 2017). Starting with the filtered CellRanger contig annotations output, combineTCR and combineExpression functions were used for combining the TCR data from each sample and for integration of the combined TCR data with the single cell RNA-seq data (processed with Seurat v.4.1.0 (ref. 66)), respectively. Repertoire overlap between the samples was quantified as the Morisita index82 with the clonalOverlap function of scRepertoire. CDR3 aa sequences previously reported in the ImmuneCODE28 and VDJdb29 databases and four recently published manuscripts31,32,33,35 were matched to the CDR3α or β sequences in the data to identify SARS-CoV-2-specific cells. The logo plots and sequence alignment plots were generated using M-Coffee83, respectively.
Tissue processing and staining for multiplexed imaging
The 5-µm tissue sections were cut from FFPE samples and placed onto glass slides. Following sectioning, glass slides (with tissue) were baked in a 60 °C oven for 1 h Deparaffinization was performed as described previously84: two exchanges of 100% xylene (10 min per exchange) followed by 100% ethanol for 10 min, 95% ethanol for 10 min, 70% ethanol for 5 min and 10% formalin for 15 min. Antigen retrieval was performed by incubating slides in AR6 buffer (Akoya Biosciences) for 40 min in a 95 °C water bath. After 40 min, slides were removed from the water bath and allowed to cool on the bench for 20 min. Sections were permeabilized, blocked and stained in PBS containing 0.3% Triton X-100 (Sigma-Aldrich), 1% bovine serum albumin (Sigma-Aldrich) and 1% human Fc block (BD Biosciences). Immunolabeling was performed with the PELCO BioWave Pro 36500-230 microwave equipped with a PELCO SteadyTemp Pro 50062 Thermoelectric Recirculating Chiller (Ted Pella) using a 2-1-2-1-2-1-2-1-2 program84,85. A complete list of antibodies and imaging panels with labeling steps can be found in Supplementary Table 10. In general, primary antibodies were applied first, washed three times in PBS and incubated with appropriate secondary antibodies. Directly conjugated primary antibodies were applied last after blocking with host serum (5%). Endogenous biotin was blocked using the Avidin/Biotin Blocking kit (Abcam). Cell nuclei were visualized with Hoechst (Biotium) and sections were mounted using Fluoromount G (Southern Biotech).
Confocal microscopy, image analysis and histocytometry
Images were acquired using an inverted Leica TCS SP8 X confocal microscope equipped with a ×40 objective (NA 1.3), 4 HyD and 1 PMT detectors, a white light laser that produces a continuous spectral output between 470 and 670 nm as well as 405, 685 and 730 nm lasers. All images were captured at an eight-bit depth, with a line average of 3 and 1,024 × 1,024 format with the following pixel dimensions: x (0.284 µm), y (0.284 µm) and z (1 µm). Images from whole-tissue sections were tiled and merged using the LAS X Navigator software (v.3.5.5.19976). Fluorophore emission was collected on separate detectors with sequential laser excitation of compatible fluorophores (3–4 per sequential) used to minimize spectral spillover. The Channel Dye Separation module within the LAS X v.3.5.5.19976 (Leica) was then used to correct for any residual spillover. Threshold identification, voxel gating, surface creation and masking were performed as previously described using Imaris software (Imaris v.9.8.0, Bitplane AG)86,87. For publication quality images, Gaussian filters, brightness/contrast adjustments and channel masks were applied uniformly to all images.
A combination of automatic and manual surface/contour creation methods were used to define GC regions of interest (ROIs) with Imaris software (Imaris v.9.8.0, Bitplane AG). GCs were identified as aggregations of five or more Ki-67+ nuclei. For each sample, whole-tissue ROIs were generated using the Hoechst channel and surface function of Imaris. The resulting metric, total area of tissue imaged, was then used to normalize the number and size of GCs between samples. Imaging data were exported and processed in Excel (Microsoft) and GraphPad Prism v.8.2.1.
Activated induced marker assay
Banked frozen PBMC and tonsil and adenoid cells were thawed as described above in ‘Processing for CITE-seq.’ Two million mononuclear cells from tonsil or adenoid or 1 million PBMCs from each donor were cultured in a 96-well round bottom plate at a concentration of 1 × 107 cells ml−1 in medium consisting of RPMI plus 5% human AB serum (Omega), 2 mM l-glutamine, 0.055 mM β-mercaptoethanol, 1% penicillin/streptomycin, 1 mM sodium pyruvate, 10 mM HEPES and 1% non-essential amino acids. Cells were blocked at 37 °C for 15 min before peptide pool stimulation with 0.5 μg ml−1 of anti-CD40 monoclonal antibody (Miltenyi). Following this, cells were stimulated with SARS-CoV-2 peptide pools for 18 h at 37 °C in 5% CO2 incubator. The following peptide pools were reconstituted per instructions and used for stimulation (Miltenyi): PepTivator SARS-CoV-2 Prot_S+, PepTivator SARS-CoV-2 Prot_S1, PepTivator SARS-CoV-2 Prot_S, PepTivator SARS-CoV-2 Prot_N, PepTivator SARS-CoV-2 Prot_M. Prot_S+, Prot_S1 and Prot_S were pooled into one megapool of spike peptides at concentration of 0.6 nmol ml−1 for each pool. PHA-L (Millipore) at 5 μg ml−1 was used as positive control. Negative control wells lacking peptides were supplemented with an equivalent volume of DMSO and ddH2O. After stimulation, cells were first stained with a viability dye (LIVE/DEAD Blue, Thermo Fisher) for 15 min at RT, washed twice and then incubated with True-Stain Monocyte Blocker (BioLegend) for 5 min. Antibodies for chemokine receptors (anti-CXCR3 for 10 min, anti-CCR7 for 10 min, anti-CXCR5 and anti-CCR6 together for 5 min) were sequentially added at RT. The antibody cocktail containing the rest of the surface antibodies and Brilliant Stain Buffer Plus (BD) was then added directly to the cells and incubated for 30 min at RT in the dark (total staining volume 180 μl). Stained cells were washed three times and fixed in 1% paraformaldehyde for 20 min at RT before collecting on the Aurora spectral cytometer (Cytek). Antibodies and reagents used in this assay are listed in Supplementary Table 10.
T cell functional assays: intracellular cytokine staining
Frozen cells were thawed as described in ‘Processing for CITE-seq.’ Two million PBMCs, adenoid or tonsil cells from each sample were resuspended in 200 μl of complete RPMI medium containing 10% FBS (VWR), 2 mM glutamine, 0.055 mM β-mercaptoethanol, 1% penicillin/streptomycin, 1 mM sodium pyruvate, 10 mM HEPES and 1% non-essential amino acids. Cells were stimulated with PMA (50 ng ml−1, Sigma) and ionomycin (1,000 ng ml−1, Sigma) for 2.5 h in the presence of anti-CD107a (BioLegend), GolgiSTOP (monensin, BD) and GolgiPlug (BFA, BD). After stimulation, surface markers were stained as described above in the AIM assay. Surface-stained cells were washed and fixed with Cytofix Fixation Buffer (BD) at RT for 20 min and washed with permeabilization buffer (eBioscience) twice. Then, the intracellular cytokine antibody mix was added for 30 min at RT (staining volume 50 μl). Stained cells were collected on the Aurora spectral cytometer (Cytek). Antibodies used in this assay are listed in Supplementary Table 10.
Viral quantification in FFPE blocks by ddPCR
RNA was extracted from scrolls cut from FFPE tonsil and adenoid tissues using the RNeasy FFPE kit (QIAGEN) according to the manufacturer’s protocol. A NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific) was used to quantify RNA concentrations. The QX200 AutoDG Droplet Digital PCR System (Bio-Rad) was used to detect and quantify SARS-CoV-2 RNA using the SARS-CoV-2 Droplet Digital PCR kit (Bio-Rad), which contains a triplex assay of primers/probes aligned to the CDC markers for SARS-CoV-2 N1 and N2 genes and human RPP30 gene. Ninety-six-well plates were prepared with technical replicates using the aforementioned kit according to the manufacturer’s instructions. The QX200 Automated Droplet Generator (Bio-Rad) provided microdroplet generation and plates were sealed with the PX1 PCR Plate Sealer (Bio-Rad) before proceeding with RT–PCR on the C1000 Touch Thermal Cycler (Bio-Rad) according to the manufacturer’s instructions. Plates were read on the QX200 Droplet Reader (Bio-Rad) and analyzed using the freely available QuantaSoft Analysis Pro Software (Bio-Rad) to quantify copies of N1, N2 and RP genes per well, which was then normalized to RNA concentration input. For samples to be considered positive for SARS-CoV-2 N1 or N2 genes, they needed to average the manufacturer’s limit of detection of ≥0.1 copies per µl and two positive droplets per well.
Statistics and reproducibility
Previous sections provide a detailed description of statistical analysis of results from unsupervised analysis as well as where to find reproducible scripts. SPICE software (v.6, NIAID, NIH, https://niaid.github.io/spice/) was used to analyze flow cytometry data on T cell polyfunctionality25. Graphs were produced by Prism (v.8). Statistical analyses were performed using SPSS (IBM, v.28.0.0.0). We did not assume that the data were normally distributed and used nonparametric statistical tests. Differences between groups were compared using the Mann–Whitney U-test for independent values and Wilcoxon signed-ranks test for paired values. Correlations were assessed using the Spearman’s rank correlation and visualized by corrplot (v.0.92). All statistical tests were two-sided. P < 0.05 was considered significant. Experiments were not repeated independently. Data collection and analysis were not performed blind to the conditions of the experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data are deposited to the Gene Expression Omnibus under accession no. GSE215802. All other source data are provided with the article or upon request from the corresponding authors. Source data are provided with this paper.
Code availability
The R scripts used in this paper are available at https://github.com/kalpanamanthiram/Covid-Tonsil.
References
Weisberg, S. P. et al. Distinct antibody responses to SARS-CoV-2 in children and adults across the COVID-19 clinical spectrum. Nat. Immunol. 22, 25–31 (2021).
Pierce, C. A. et al. Immune responses to SARS-CoV-2 infection in hospitalized pediatric and adult patients. Sci. Transl. Med. 12, eabd5487 (2020).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Huang, N. et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat. Med. 27, 892–903 (2021).
Wagar, L. E. et al. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 27, 125–135 (2021).
Kumar, B. V. et al. Human tissue-resident memory T Cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20, 2921–2934 (2017).
Mitchell, R. B. et al. Clinical practice guideline: tonsillectomy in children (update). Otolaryngol. Head Neck Surg. 160, S1–S42 (2019).
Korber, B. et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827 (2020).
Chan, W. S. et al. Geographical prevalence of SARS-CoV-2 variants, August 2020 to July 2021. Sci. Rep. 12, 4704 (2022).
Poon, M. M. L. et al. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci. Immunol. 6, eabl9105 (2021).
Kaneko, N. et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell 183, 143–157 (2020).
Woodruff, M. C. et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 21, 1506–1516 (2020).
Sokal, A. et al. Maturation and persistence of the anti-SARS-CoV-2 memory B cell response. Cell 184, 1201–1213 (2021).
Attaf, N. et al. FB5P-seq: FACS-based 5-prime end single-cell RNA-seq for integrative analysis of transcriptome and antigen receptor repertoire in B and T Cells. Front. Immunol. 11, 216 (2020).
Albrecht, I. et al. Persistence of effector memory TH1 cells is regulated by Hopx. Eur. J. Immunol. 40, 2993–3006 (2010).
Ben Mkaddem, S., Benhamou, M. & Monteiro, R. C. Understanding Fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front. Immunol. 10, 811 (2019).
Raybould, M. I. J., Kovaltsuk, A., Marks, C. & Deane, C. M. CoV-AbDab: the coronavirus antibody database. Bioinformatics 37, 734–735 (2021).
Ding, Y., Yan, H. & Guo, W. Clinical characteristics of children with COVID-19: a meta-analysis. Front. Pediatrics 8, 431 (2020).
Kim, C. H. et al. Subspecialization of CXCR5+ T cells: B helper activity is focused in eagerminal center-localized subset of CXCR5+ T cells. J Exp Med. https://pubmed.ncbi.nlm.nih.gov/11413192/(2001).
Padhan, K., Moysi, E., Noto, A. & Petrovas, C. Acquisition of optimal TFH cell function is defined by specific molecular, positional, and TCR dynamic signatures. Proc. Natl Acad. Sci. USA https://www.pnas.org/doi/full/10.1073/pnas.2016855118 (2021).
Li, H. & Pauza, C. D. CD25+Bcl6low T follicular helper cells provide help to maturing B cells in germinal centers of human tonsil. Eur. J. Immunol. 45, 298–308 (2015).
Mahnke, Y. D., Brodie, T. M., Sallusto, F., Roederer, M. & Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 43, 2797–2809 (2013).
Piedra-Quintero, Z. L., Wilson, Z., Nava, P. & Guerau-de-Arellano, M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front. Immunol. 11, 597959 (2020).
Jung, J. H. et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat. Commun. 12, 1–12 (2021).
De Biasi, S. et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 11, 3434 (2020).
Adamo, S. et al. Signature of long-lived memory CD8+ T cells in acute SARS-CoV-2 infection. Nature 602, 148–155 (2022).
Saini, S. K. et al. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8+ T cell activation in COVID-19 patients. Sci. Immunol. 6, eabf7550 (2021).
Nolan, S. et al. A large-scale database of T-cell receptor β (TCRβ) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Res. Sq. https://doi.org/10.21203/rs.3.rs-51964/v1 (2020).
Bagaev, D. V. et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 48, D1057–D1062 (2019).
Grifoni, A. et al. SARS-CoV-2 human T cell epitopes: adaptive immune response against COVID-19. Cell Host Microbe 29, 1076–1092 (2021).
Mudd, P. A. et al. SARS-CoV-2 mRNA vaccination elicits a robust and persistent T follicular helper cell response in humans. Cell 185, 603–613 (2022).
Bacher, P. et al. Low-avidity CD4(+) T cell responses to SARS-CoV-2 in unexposed individuals and humans with severe COVID-19. Immunity 53, 1258–1271 (2020).
Meckiff, B. J. et al. Imbalance of regulatory and cytotoxic SARS-CoV-2-reactive CD4(+) T cells in COVID-19. Cell 183, 1340–1353 (2020).
Al Balushi, A. et al. Immunological predictors of disease severity in patients with COVID-19. Int. J. Infect. Dis. 110, 83–92 (2021).
Lu, X. et al. Identification of conserved SARS-CoV-2 spike epitopes that expand public cTFH clonotypes in mild COVID-19 patients. J. Exp. Med. 218, e20211327 (2021).
Mudd, P. et al. Examining multi-level immune response to determine prevalence of COVID-19 in pediatric tonsillectomy. Laryngoscope https://doi.org/10.1002/lary.30382 (2022).
Files, J. K. et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J. Clin. Investig. 131, e140491 (2021).
Gaebler, C. et al. Evolution of antibody immunity to SARS-CoV-2. Nature 591, 639–644 (2021).
Breton, G. et al. Persistent cellular immunity to SARS-CoV-2 infection. J. Exp. Med. 218, e20202515 (2021).
Roukens, A. H. E. et al. Prolonged activation of nasal immune cell populations and development of tissue-resident SARS-CoV-2-specific CD8+ T cell responses following COVID-19. Nat. Immunol. 23, 23–32 (2022).
Smith, N. et al. Distinct systemic and mucosal immune responses during acute SARS-CoV-2 infection. Nat. Immunol. 22, 1428–1439 (2021).
Szabo, P. A., Miron, M. & Farber, D. L. Location, location, location: tissue-resident memory T cells in mice and humans. Sci. Immunol. 4, eaas9673 (2019).
Yoshida, M. et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature 602, 321–327 (2022).
He, R. et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–416 (2016).
Leong, Y. A. et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 17, 1187–1196 (2016).
Yu, D. & Ye, L. A portrait of CXCR5+ follicular cytotoxic CD8+ T cells. Trends Immunol. 39, 965–979 (2018).
Focosi, D., Bestagno, M., Burrone, O. & Petrini, M. CD57+ T lymphocytes and functional immune deficiency. J. Leukoc. Biol. 87, 107–116 (2010).
Kim, T. S., Hufford, M. M., Sun, J., Fu, Y. X. & Braciale, T. J. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J. Exp. Med. 207, 1161–1172 (2010).
Chertow, D. et al. SARS-CoV-2 infection and persistence throughout the human body and brain. Nat. Portfolio https://doi.org/10.21203/rs.3.rs-1139035/v1 (2022).
Tan, H.-X. et al. Cutting edge: SARS-CoV-2 infection induces robust germinal center activity in the human tonsil. J. Immunol. 208, 2267–2271 (2022).
Seay, H. R. et al. Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 1, e88242 (2016).
Bergqvist, P. et al. Re-utilization of germinal centers in multiple Peyer’s patches results in highly synchronized, oligoclonal, and affinity-matured gut IgA responses. Mucosal Immunol. 6, 122–135 (2013).
Meng, W. et al. An atlas of B-cell clonal distribution in the human body. Nat. Biotechnol. 35, 879–884 (2017).
Ohm-Laursen, L. et al. B cell mobilization, dissemination, fine tuning of local antigen specificity and isotype selection in asthma. Front. Immunol. 12, 702074 (2021).
Boyaka, P. N. et al. Human nasopharyngeal-associated lymphoreticular tissues. Functional analysis of subepithelial and intraepithelial B and T cells from adenoids and tonsils. Am. J. Pathol. 157, 2023–2035 (2000).
Feldstein, L. R. et al. Characteristics and outcomes of US children and adolescents with multisystem inflammatory syndrome in children (MIS-C) compared with severe acute COVID-19. JAMA 325, 1074–1087 (2021).
Sacco, K. et al. Immunopathological signatures in multisystem inflammatory syndrome in children and pediatric COVID-19. Nat. Med. 28, 1050–1062 (2022).
Michael, S. et al. Standardization of ELISA protocols for serosurveys of the SARS-CoV-2 pandemic using clinical and at-home blood sampling. Nat. Commun. 12, 113 (2021).
Kalish, H. et al. Undiagnosed SARS-CoV-2 seropositivity during the first 6 months of the COVID-19 pandemic in the United States. Sci. Transl. Med. 13, eabh3826 (2021).
Zahra, F. T., Bellusci, L., Grubbs, G., Golding, H. & Khurana, S. Neutralisation of circulating SARS-CoV-2 delta and omicron variants by convalescent plasma and SARS-CoV-2 hyperimmune intravenous human immunoglobulins for treatment of COVID-19. Ann. Rheum. Dis. https://doi.org/10.1136/annrheumdis-2022-222115 (2022).
Ravichandran, S. et al. Antibody signature induced by SARS-CoV-2 spike protein immunogens in rabbits. Sci. Transl. Med. 12, abc3539 (2020).
Tang, J. et al. Antibody affinity maturation and plasma IgA associate with clinical outcome in hospitalized COVID-19 patients. Nat. Commun. 12, 1221 (2021).
Park, L. M., Lannigan, J. & Jaimes, M. C. OMIP-069: forty-color full spectrum flow cytometry panel for deep immunophenotyping of major cell subsets in human peripheral blood. Cytom. A 97, 1044–1051 (2020).
Liu, C. et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 184, 1836–1857 (2021).
Kotliarov, Y. et al. Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat. Med. 26, 618–629 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Mulè, M. P., Martins, A. J. & Tsang, J. S. Normalizing and denoising protein expression data from droplet-based single cell profiling. Preprint at bioRxiv https://doi.org/10.1101/2020.02.24.963603 (2021).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. 1st edn, (Springer, 2009).
Giudicelli, V., Chaume, D. & Lefranc, M. P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 33, D256–D261 (2005).
Ye, J., Ma, N., Madden, T. L. & Ostell, J. M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41, W34–W40 (2013).
Gupta, N. T. et al. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 (2015).
Nouri, N. & Kleinstein, S. H. A spectral clustering-based method for identifying clones from high-throughput B cell repertoire sequencing data. Bioinformatics 34, i341–i349 (2018).
Gupta, N. T. et al. Hierarchical clustering can identify B cell clones with high confidence in Ig repertoire sequencing data. J. Immunol. 198, 2489–2499 (2017).
Yaari, G. et al. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front. Immunol. 4, 358 (2013).
Hoehn, K. B., Pybus, O. G. & Kleinstein, S. H. Phylogenetic analysis of migration, differentiation, and class switching in B cells. PLoS Comput. Biol. 18, e1009885 (2022).
Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots v.0.4.0. https://github.com/kassambara/ggpubr (2020).
Hoehn, K. B. et al. Repertoire-wide phylogenetic models of B cell molecular evolution reveal evolutionary signatures of aging and vaccination. Proc. Natl Acad. Sci. USA 116, 22664–22672 (2019).
Nielsen, R. & Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–936 (1998).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Borcherding, N., Bormann, N. L. & Kraus, G. scRepertoire: an R-based toolkit for single-cell immune receptor analysis. F1000Res 9, 47 (2020).
Greiff, V., Miho, E., Menzel, U. & Reddy, S. T. Bioinformatic and statistical analysis of adaptive immune repertoires. Trends Immunol. 36, 738–749 (2015).
Wallace, I. M., O’Sullivan, O., Higgins, D. G. & Notredame, C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34, 1692–1699 (2006).
Radtke, A. J. et al. IBEX: an iterative immunolabeling and chemical bleaching method for high-content imaging of diverse tissues. Nat. Protoc. 17, 378–401 (2022).
Radtke, A. J. et al. IBEX: a versatile multiplex optical imaging approach for deep phenotyping and spatial analysis of cells in complex tissues. Proc. Natl Acad. Sci. USA 117, 33455–33465 (2020).
Radtke, A. J. et al. Lymph-node resident CD8α+ dendritic cells capture antigens from migratory malaria sporozoites and induce CD8+ T cell responses. PLoS Pathog. 11, e1004637 (2015).
Gerner, M. Y., Kastenmuller, W., Ifrim, I., Kabat, J. & Germain, R. N. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376 (2012).
Acknowledgements
We thank the patients and their families for their generous participation; J. Reilley and N. Bansal for their technical assistance; A.J. Athman, R. Kissinger, R. Perry-Gottschalk and A. Stewart of the Research Technologies Branch of the National Institute of Allergy and Infectious Diseases (NIAID) for figure illustrations and formatting; the Division of Otolaryngology at CNH for helping with participant recruitment; the National Cancer Institute (NCI) Sequencing Facility for sequencing support; J. Lack, J. Cannons, A. Pichler, A.I. Lim, L. Notarangelo and Y. Belkaid (NIAID/National Institutes of Health (NIH)), S. Anderson and D. Kastner (National Human Genome Research Institute (NHGRI)/NIH) and K. Edwards (Vanderbilt) for insightful discussions. This work was supported in part by the Intramural Research Programs of NIAID, Clinical Center, NHGRI, NCI and National Institute of Biomedical Imaging and Bioengineering at NIH that supported individual investigators and the Intramural Research Programs of NIAID and other NIH Institutes that support the NIH Center for Human Immunology. The antibody response study was supported by the US Food and Drug Administration’s Perinatal Health Center of Excellence project grant no. GCBER005 to S.K. K.B.H. and S.H.K. were funded in part by NIAID/NIH grant no. R01AI104739. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
K.M., P.M., P.L.S. and Q.X. conceived and designed the study. Q.X., K.M., A.J.R., J.C., J.T., G.G., S.S., S.R., J.K., M. Karkanitsa, J.S., H.K., M.M.K., M. Kirby, F.C., G.K., P.D. and G.S. performed experiments. Q.X, K.M., P.M.-A., A.J.M., A.J.R., K.B.H., C.O., C.L., J.K., S.S., S.R., F.C., A.S. and I.T.M. analyzed and interpreted results. K.M., P.M., H.B., N.R., D.P. and L.G. developed patient recruitment materials and/or recruited participants. Q.X., P.M.-A., A.J.M., A.J.R., K.B.H., L.K., S. Preite, R.A., P.J.M., D.M.S., S. Pittaluga, R.N.G., S.M., D.S.C., A.S., S.K., S.H.K., K.S., J.S.T., P.M., P.L.S. and K.M. provided critical scientific input and/or reagents. K.M., P.L.S. and Q.X. wrote the initial draft of the paper. All authors contributed to the final review and editing of the paper.
Corresponding authors
Ethics declarations
Competing interests
P.M.-A. is currently an employee of Novartis. S. Preite and A.S. are currently employees of AstraZeneca and may own stock or stock options. S.H.K. receives consulting fees from Peraton. K.B.H. receives consulting fees from Prellis Biologics. The remaining authors have no competing interests.
Peer review
Peer review information
Nature Immunology thanks Olivier Schwartz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ioana Visan in collaboration with the Nature Immunology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Characterization of neutralization titers and S1+RBD+ B cells.
a. Correlation of S1+RBD+ cell frequency among B cells in post-COV PBMCs, tonsils and adenoids. Data point color indicates neutralizing titers (PsVNA50) to WA-1. Donors with the lowest frequencies of S1+RBD+ B cells are labeled. Spearman’s coefficient (r) noted. b. CD27+ BSM cell frequency among total B cells and among S1+RBD+ B cells from post-COV adenoids (p < 0.0001) and tonsils (p < 0.0001). c. S1+RBD+ B cell frequency among CD27+ BSM cells in post-COV adenoids and tonsils according to time from positive PCR/antigen test to surgery. d. Proportion of each isotype among S1+RBD+ CD27+ BSM cells and total CD27+ BSM cells in post-COV PBMCs, adenoids and tonsils. e. Percentage of S1+RBD+ B cells among CD27+ BSM cells from post-COV and UC PBMCs, adenoids and tonsils (all post-COV vs. UC p < 10−6). f,g Mean number of GCs per total scanned tissue area (f) and mean GC area (total GC area/number of GCs in section) (g) from adenoids and tonsils from post-COV and UC donors (n = 3 each). h,i. Percentage of S1+RBD+ B cells among GC B cells (h) and total B cells (i) from 14 pairs of post-COV adenoids and tonsils (total B p = 0.007, GC B p = 0.030) and UC (UC adenoid n = 27; tonsil n = 30). All post-COV vs. UC comparisons p < 10−6. j. Summary of correlations between frequencies of S1+RBD+ cells among CD19+ B cells PBMCs, adenoids and tonsils and neutralizing titers (PsVNA50) to multiple variants. Spearman’s correlation noted in color. % S1+RBD+ B in post-COV adenoid vs. PBMC p = 0.006, tonsil vs. PBMC p = 0.00003, tonsil vs. adenoid p = 0.0003; % S1+RBD+ B in post-COV adenoid vs. PsVNA50 Beta p = 0.01, Iota p = 0.04, Delta p = 0.05, Omicron p = 0.04). Panels a–g,j: PBMC post-COV n = 18, UC n = 33; adenoid post-COV n = 16, UC n = 27; and tonsil post-COV n = 16, UC n = 30. Each symbol represents one donor. Means ± S.D. displayed in bar plots. Significance calculated with two-sided Mann–Whitney U-test (unpaired) or Wilcoxon signed ranks test (paired). *p < 0.05, ***p < 0.001, ****p < 0.0001.
Extended Data Fig. 2 CITE-seq analysis of SARS-CoV-2 antigen-specific B cells.
a,b. Heat map of unsupervised clustering by CITE-seq antibody expression of S1+ and S1− B cells from tonsils, adenoids and PBMCs from three donors (2 post-COV and 1 UC) yielding 15 clusters (a). Expression of signature gene sets for GC B cells, memory B (Mem) cells and plasma cells/plasmablasts (PC/PB) among all B cells (S1+ and S1−) organized by cluster (b). c. Heat map showing differentially expressed (DE) genes in S1+ vs. S1− B cells from tonsils and adenoids from cluster 2 (which are CD27+ BSM cells), see Supplementary Table 5. d. Sub-isotype percentages among sorted S1+ and S1− B cells from adenoids, tonsils and PBMCs of 2 post-COV donors (CNMC71 and 89) and one UC (CNMC99). Raw number of cells with a given sub-isotype are labeled only for sub-isotypes that make up >10% of a given category. e. Somatic hypermutation (SHM) frequency (calculated in V gene) among sorted S1+ and S1− B cells of all isotypes from PBMCs, adenoids and tonsils of each donor. Median ± quartiles and p values shown in plots. Significance calculated with two-sided Mann–Whitney U-test. CNMC71 PBMC S1+ n = 101, S1− n = 577 cells; CNMC89 PBMC S1+ n = 44, S1− n = 1491 cells; CNMC99 PBMC S1− n = 1026 cells; CNMC71 adenoid S1+ n = 191, S1− n = 1177 cells; CNMC89 adenoid S1+ n = 261, S1− n = 1647 cells; CNMC99 adenoid S1+ n = 40, S1− n = 1593 cells; CNMC71 tonsil S1+ n = 286, S1− n = 1514 cells; CNMC89 tonsil S1+ n = 416, S1− n = 2644 cells; CNMC99 tonsil S1+ n = 66, S1− n = 2346 cells. f. Sub-isotype frequencies among S1+ B cells from clones shared between tonsil and adenoid and unshared clones. Raw number of cells with a given sub-isotype are labeled only for sub-isotypes that make up >10% of a given category.
Extended Data Fig. 3 UMAP of unsupervised clustering of B cells from tonsil and adenoid.
a. Uniform manifold approximation and projection (UMAP) of unsupervised clustering of surface markers from flow cytometric analysis of CD19+ B cells from adenoids and tonsils. b. Heatmaps of marker/antibody expression overlayed on UMAP.
Extended Data Fig. 4 UMAP of unsupervised clustering of CD4+ T cells from tonsil and adenoid.
a. Comparison of CD3+, CD4+ and CD8+ T cell frequency in adenoid of post-COV (n = 17) and UC donors (n = 42), CD3+ p value = 0.043, CD4+ p = 0.017. b,c. UMAP of unsupervised clustering of surface markers from flow cytometric analysis of CD4+ T cells from adenoid and tonsil (b) with heatmaps of marker/antibody expression overlayed (c).
Extended Data Fig. 5 Phenotyping of expanded CD4+ T cell populations.
a,b. Correlation between frequency of CD57+PD-1hi CD4+ T cells and frequency of GC B cells in adenoids (a, n = 59) and tonsils (b, n = 64). c,d. Intracellular cytokine and cytotoxic factor expression in various CD4+ T cell subsets gated on CD57 and PD-1 from post-COV adenoids (c, n = 13) and tonsils (d, n = 13) after PMA/ionomycin stimulation. Mean cell frequency shown in heat map. e. Correlations among various subsets of SARS-CoV-2-specific B cells (defined in Supplementary Figs. 1) and significantly different tissue CD4+ T cell clusters (clusters 3, 6, 9 shown as % of CD4+ T cells) from unsupervised analysis. f. Percentage of CD25+CXCR5+PD-1hi cells among CD4+ T cells in post-COV and UC adenoids and tonsils (p = 0.031). g. Cytokine production by CD25+ and CD25− CXCR5+PD-1hi CD4+ T cells in tonsils (n = 26) and adenoids (n = 26) following PMA/ionomycin stimulation, all p < 0.0001. h. Correlation between frequency of CD25+CXCR5+PD-1hi CD4+ T cell and GC B cell frequencies in tonsils (n = 64). i. Frequency of CXCR3+CCR6− cells among pre-TFH cells (PD-1intCXCR5+ conventional CD4+ T) in post-COV and UC adenoids (p = 0.042) and tonsils. j,k. Intracellular cytokine/cytotoxic factor expression in different pre-TFH cell subsets gated on CXCR3 and CCR6 from post-COV adenoids (j, n = 13) and tonsils (k, n = 13) after PMA/ionomycin stimulation. Mean cell frequency shown in heat map. l. Comparison of IFN-γ production by CD4+ T cells in adenoids and tonsils following PMA/ionomycin stimulation (n = 26 including 13 post-COV and 13 UC samples of each tissue, p < 0.0001). For panels, f, and i: adenoids post-COV n = 17, UC n = 42; tonsils post-COV n = 18, UC n = 46. Each symbol represents one donor. Means ± S.D. displayed on bar plots. Significance calculated with two-sided Mann–Whitney U-test to compare two groups and Spearman’s rank test for correlations (r is Spearman’s coefficient). *p < 0.05, ****p < 0.0001.
Extended Data Fig. 6 cTFH cell populations are expanded post-COVID-19 in PBMC.
a,b. UMAP of unsupervised clustering of surface markers from flow cytometric analysis of CD4+ T cells from PBMCs (a) with heatmaps of marker/antibody expression overlayed (b). c. Intracellular cytokine and cytotoxic factor production by various circulating TFH cell (cTFH) subsets in PBMC gated by CXCR3 and CCR6 from post-COV donors (n = 4) following PMA/ionomycin stimulation. Mean cell frequency shown in heat map. d. Frequency of CD45RA+CCR7+CD28+CD27+CD95+ CD4+ T stem cell-like memory (TSCM) cells in PBMC of post-COV (n = 16) and UC (n = 41), p = 0.007. Each symbol represents one donor. Means ± S.D. displayed on bar plots. Significance calculated with two-sided Mann–Whitney U-test. **p < 0.01.
Extended Data Fig. 7 UMAP of unsupervised clustering of CD8+ T cells from tonsil and adenoid.
a. UMAP of unsupervised clustering of surface markers from flow cytometric analysis of CD8+ T cells from adenoids and tonsils. b. Heatmaps of marker/antibody expression overlayed on UMAP.
Extended Data Fig. 8 Expanded CD8+ T cell populations after COVID-19.
a. Quantification of the effect of previous SARS-CoV-2 infection on CD8+ T cell clusters in tonsil showing regression coefficients ± 95% confidence intervals (CI) and p values, estimated with a linear model controlling for age and sex (post-COV n = 15, UC n = 42). b,c. Frequencies of CD45RA+CCR7+CD8+ naïve T (TN) and CD45RA−CCR7−CD8+ effector memory T (TEM) cells in post-COV and UC adenoids (b) and tonsils (c, p = 0.035 for TEM). d. Frequency of CXCR3+CCR6− cells among CD8+ T cells in post-COV and UC adenoids (p = 0.022) and tonsils. e. Comparison of IFN-γ production by CD8+ T cells in adenoids and tonsils following PMA/ionomycin stimulation (n = 26 for each tissue, p = 0.003). f,g. Intracellular cytokine/cytotoxic factor production by different CD8+ T cell subsets gated by CD57 and PD-1 from post-COV adenoids (f, n = 13) and tonsils (g, n = 13). Mean cell frequency shown in heat map. h. Unsupervised clustering of CD8+ T cells from PBMCs according to surface antibodies from flow cytometric analysis. No clusters showed significant differences (p < 0.05) in post-COV (n = 13) and UC (n = 34) samples. i. Quantification of the effect of previous SARS-CoV-2 infection on CD8+ T cell clusters in PBMCs showing regression coefficients ± 95% CI and p values, estimated with a linear model controlling for age and sex. j. Frequency of CD45RA+CCR7+CD28+CD27+CD95+ CD8+ T stem cell-like memory (TSCM) in post-COV (n = 16) and UC (n = 41) PBMCs (p = 0.002). For panels b–d, adenoids post-COV n = 17, UC n = 42, tonsils post-COV n = 18, UC n = 46. Each symbol represents one donor. Means ± S.D. displayed on bar plots. Significance calculated with two-sided Mann–Whitney U-test. *p < 0.05, **p < 0.01.
Extended Data Fig. 9 SARS-CoV-2 antigen-specific T cells and TCR repertoire.
a,b. Frequencies of AIM+ (OX40+4-1BB+) CD4+ T cells from adenoid (a) and tonsil (b) of post-COV tonsils (n = 6) and adenoids (n = 6) following SARS-CoV-2 spike (S), membrane (M) and nucleocapsid (N) peptide pool stimulation. DMSO (vehicle, V) is negative control. Significance calculated with two-sided Wilcoxon signed rank test for paired samples from the same donor. c. Among expanded CD8+ T cell clones, those with TCRβ CDR3 amino acid (aa) sequences that match those publicly reported to be SARS-CoV-2-reactive are highlighted in the UMAP (clustering shown in Fig. 7h). d. Frequency of matches among expanded CD8+ T cells from two post-COV donors (CNMC71 and 89) and one UC (CNMC99). More PBMCs were sorted than tonsil or adenoid cells in order to sort similar numbers of S1+ B cells from each sample; therefore, more T cells were analyzed from PBMCs than tonsil or adenoid. e. Antigens recognized by CD8+ T cells in post-COV samples with CDR3β aa sequences publicly reported to be SARS-CoV-2 reactive; proportion of cells recognizing each antigen is shown in the pie chart. f,g. Frequency of CD4+ T cells that are part of expanded clonotypes (frequency >0.001 and absolute count ≥3) in tonsils, adenoids and PBMCs. Clones were defined by identical CDR3α (f) or CDR3β (g) aa sequences. h–m. UMAP (h), tissue distribution (i) and CITE-seq surface antibody expression (j) of 14 clusters of sorted CD95+CD4+ T cells from tonsils, adenoids and PBMCs of 2 post-COV donors and one UC. Expanded TCRα or β clonotypes (k) and distribution of expanded clones across clusters (l). Expanded TCRα clones with CDR3 sequences that match publicly reported SARS-CoV-2-specific sequences (m). n. Overlap of CD4+ T cell clones among PBMCs, tonsils and adenoids from 2 post-COV donors and one UC; degree of overlap between TCRα/β CDR3 aa sequences was calculated with the Morisita index (shown in plot), ranging from 0 to 1, with 0 indicating no sharing and 1 indicating full overlap.
Extended Data Fig. 10 Summary of findings.
Schematic illustrating the immunologic profile of the pharyngeal lymphoid tissues and peripheral blood of COVID-19-convalescent children including (1) SARS-CoV-2-specific GC B, memory B and T cells with overlapping B and CD8+ T cell clones in the tonsils and adenoids, (2) persistent changes in lymphocyte populations involved in GC and anti-viral responses, which were most prominent in the adenoid, with type 1 (IFN-γ-associated) skewing of several T lymphocyte populations, and (3) persistence of viral RNA in the tissue.
Supplementary information
Supplementary Information
Supplementary Tables 1–12 and Supplementary Figs. 1–10.
Supplementary Table 2
Participant characteristics and summary of samples used in immune profiling.
Supplementary Table 4
Serologic test summary and data.
Supplementary Table 5
DE genes S1 positive versus S1 negative B cells.
Supplementary Table 6
Publicly reported BCR matches.
Supplementary Table 7
Characteristics of shared clones.
Supplementary Table 8
TCR repertoire analysis.
Supplementary Table 9
ddPCR results from FFPE.
Supplementary Table 10
Reagents.
Supplementary Table 11
Samples used in functional analyses and imaging.
Supplementary Table 12
Unsupervised analysis of flow cytometry data – raw data.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Rights and permissions
About this article

Cite this article
Xu, Q., Milanez-Almeida, P., Martins, A.J. et al. Adaptive immune responses to SARS-CoV-2 persist in the pharyngeal lymphoid tissue of children. Nat Immunol 24, 186–199 (2023). https://doi.org/10.1038/s41590-022-01367-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-022-01367-z
This article is cited by
-
The Mouth as a Site of SARS-CoV-2 Infection
Current Oral Health Reports (2024)
-
Deep phenotyping of post-infectious myalgic encephalomyelitis/chronic fatigue syndrome
Nature Communications (2024)
-
SARS-CoV-2 reservoir in post-acute sequelae of COVID-19 (PASC)
Nature Immunology (2023)
