
Abstract
Injury is a key driver of inflammation, a critical yet necessary response involving several mediators that is aimed at restoring tissue homeostasis. Inflammation in the central nervous system can be triggered by a variety of stimuli, some intrinsic to the brain and others arising from peripheral signals. Fine-tuned regulation of this response is crucial in a system that is vulnerable due to, for example, aging and ongoing neurodegeneration. In this context, seemingly harmless interventions like a common surgery to repair a broken limb can overwhelm the immune system and become the driver of further complications such as delirium and other perioperative neurocognitive disorders. Here, we discuss potential mechanisms by which the immune system affects the central nervous system after surgical trauma. Together, these neuroimmune interactions are becoming hallmarks of and potential therapeutic targets for multiple neurologic conditions, including those affecting the perioperative space.
Similar content being viewed by others

Main
The influence of systemic factors on the central nervous system (CNS) has long been shown; however, the molecular underpinnings of these factors and their effect on brain functioning have only recently become appreciated. It is becoming more apparent that, under certain conditions such as aging and neurodegeneration, systemic inflammatory mediators can influence the brain and ultimately impact cognition1. For example, in stroke and traumatic brain injury, peripheral cells can directly enter the brain parenchyma and cause neuroinflammation, especially when the blood–brain barrier (BBB) is damaged2,3.
Surgery can be lifesaving and can also vastly improve quality of life. Over the past decade, however, growing evidence has linked surgery, such as cardiac and orthopedic procedures, to brain pathology similar to that of other neurologic diseases in which the brain is the primary target of injury. This scenario is particularly true in older adults and frail patients who already experience limited cognitive reserves and are more vulnerable to cognitive deterioration and even dementia. Indeed, perioperative neurocognitive disorders (PNDs), which include acute delirium and longer-lasting cognitive decline, are now considered some of the most common postoperative complications among older adults4. PNDs can develop following major procedures, especially cardiac and orthopedic surgery. The strongest risk factors for postoperative delirium are advanced age and dementia; a thorough review of the relevant clinical literature is provided in ref. 5. Notably, anesthesia has been implicated in the pathogenesis of PNDs, but clinically evident cognitive decline can occur after regional or general anesthesia, suggesting that other factors, such as surgical trauma, play a prominent role in causing cognitive deficits6. Indeed, orthopedic surgery is routinely performed in older adults, and as many as 50% of these patients suffer from postoperative delirium, an acute and fluctuating disturbance in awareness and attention7. Despite its acute course, typically 1–3 days after surgery and anesthesia, delirium has devastating consequences: increased 1-year postoperative mortality8,9, decreased quality of life10 and an increased long-term risk for Alzheimer’s disease (AD)11,12. Importantly, PNDs have a synergistic relationship with neurodegenerative diseases, making their impact even more concerning13. In particular, an episode of delirium can accelerate the trajectory of cognitive decline, which can contribute to further dementia14. When delirium occurs in patients with underlying dementia, the prognosis is even worse15,16. In fact, patients who suffer delirium after hip fracture surgery have a twofold increased risk for 1-year mortality as compared to patients who have had hip fracture surgery but do not have dementia or delirium17. Furthermore, delirium not only occurs after surgery but is frequently observed in the medical intensive care unit as a result of critical illness and mechanical ventilation, particularly relevant in the recent context of COVID-19, with an ~25% incidence, which is probably underestimated18,19. Delirium already contributes an estimated $150 billion per year to the soaring healthcare costs in the United States20. These complications are generating new challenges for our aging society and are rapidly becoming a significant burden for families and healthcare providers21. Preclinical and early clinical studies continue to investigate putative mechanisms for PNDs, focusing in particular on a role for neuroinflammation. Perhaps such a role is not surprising; after all, surgery is a form of controlled trauma, trauma is an established source of tissue injury, and injury is a key driver of inflammation. Here, we will review key mechanisms related to postoperative inflammation and the implications for PND development, focusing on neuroinflammation and key cellular targets affected by surgical trauma.
Systemic inflammation and DAMPs after sterile injury
The brain has long been referred to as an immune-privileged organ, although it is becoming more apparent that neuroimmune interactions between the periphery and the CNS are not rare or restricted to brain neuropathology22. The presence of a physical barrier, the BBB, supported by different cell types, including brain endothelial cells, astrocytes and pericytes, prevents direct access of damaging molecules to the CNS23. The role of the BBB in homeostasis and pathology has been extensively reviewed (see ref. 23). Here, we will focus on the emerging role of postoperative inflammation as a trigger for PND pathology.
Millions of patients worldwide undergo surgery routinely, including more invasive procedures that involve extensive tissue damage, hemorrhage and ischemic damage due to clamping or tourniquets, used in many cardiac and orthopedic procedures. These forms of sterile trauma are associated with robust systemic inflammation that, in some cases, can lead to significant complications and even death. Cellular damage after surgical trauma triggers endogenous factors known as damage-associated molecular patterns (DAMPs), which activate immune cells such as neutrophils and monocytes to resolve the damage and restore homeostasis (reviewed in detail in ref. 24). Activation of these cells contributes to systemic inflammation, which can impact multiple organs, including the brain. The role of the innate immune response after trauma has been recently reviewed in ref. 25. There is growing evidence to suggest that dysregulated immune functions are a driver of PNDs.
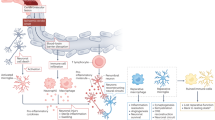
HMGB1 is a prototypical DAMP with distinct roles in trauma and infection. HMGB1 is both a nuclear factor and a secreted protein that actively regulates several inflammatory processes26. In the nucleus, it is responsible for chromatin packaging, and it can be rapidly released into the circulation following a mechanical injury such as incision of tissues or vessels27. HMGB1 is detectable in the circulation within 30 minutes after surgery28. Together with other cytokines such as tumor necrosis factor (TNF), it contributes to the early mobilization of immune cells after surgery and the subsequent engagement of bone-marrow-derived macrophages (BMDMs)29. Indeed, exogenous administration of HMGB1 itself recapitulates similar neuroimmune and cognitive deficits found in PNDs, suggesting that early release of DAMPs jumpstarts the neuroinflammatory and behavioral responses to trauma. Soluble HMGB1 engages several pattern recognition receptors (PRRs), including Toll-like receptor (TLR) 2 and TLR4 as well as the receptor RAGE30. Both TLR4 and MyD88 signaling have been implicated in PND pathology, as evidenced by mice genetically deficient for both genes that remain protected from surgery-induced inflammation and CNS dysfunction28,31. Indeed, HMGB1 is a potent primer for CNS inflammation. Administration of antibodies selective for this DAMP can ‘desensitize’ resident microglia to peripheral inflammation caused by both infective and sterile trauma, including in models of PND29,32,33. HMGB1 signaling can also interact with extracellular ATP, also commonly released after tissue/cellular trauma, and together, they can activate the inflammasome complex to release interleukin (IL)-1β and IL-1834. Although the role of IL-1β in PNDs has been established through both preclinical models and the biofluids of postoperative patients35,36, the regulation and characterization of the cellular source(s) of IL-1 remain poorly described. A study described a key role for P2X7 receptor signaling in postoperative neuroinflammation, suggesting that blocking ATP binding to this receptor prevents inflammasome activation and improves cognitive outcomes after surgery (Fig. 1)37.

Aseptic surgical trauma triggers acute inflammation by inducing inflammatory cytokines and DAMPs. This inflammatory milieu contributes to the recruitment of immune cells at the site of injury, for example, the tibia following intramedullary pinning and fixation, but also affects functions in other organs, including the brain. Monocyte activation is one of the drivers for postoperative neuroinflammation, as found in both preclinical and clinical PND studies. Key PRRs expressed on the surface of monocytes have been implicated in this signaling. HMGB1 binds to TLR4 and RAGE to activate NF-κB, which further transcribes de novo cytokines. This process can synergize with the activation of the NLRP3 inflammasome complex to further elevate the production of IL-1. Plasma and CSF levels of IL-1β have been described in individuals with PNDs. IL-1 induces expression of MCP-1 and facilitates access of CCR2+ monocytes in the CNS, possibly contributing to a feed-forward loop leading to non-resolving inflammation and neurologic pathology. NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells.
Postoperative complement system activation
The complement system is another key component in the inflammatory response, which can be activated by the ‘DAMPs surge’ after surgery. For example, C-reactive protein (CRP), a biomarker that has been well established by studies involving multiple cohorts of patients with delirium, can activate and regulate the classical complement pathway38,39,40,41. While CRP is commonly used clinically as a biomarker, less attention has been devoted to the complement cascade and its potential implications for PNDs. Previous clinical investigations demonstrated that in patients who were postoperative or had multiple traumas, there is early complement component 3 (C3) activation, represented by plasma C3 depletion and upregulation of cleaved forms of C3 (including C3a and C3b)42,43,44. In a murine model of orthopedic surgery, C3 was upregulated in the CNS, with higher expression of C3 in hippocampal astrocytes and C3a receptor specifically in microglia45. Although C3 and C3a concentrations were not measured in the plasma, the study showed that administration of a C3a receptor blocker improved choroidal blood–cerebrospinal fluid barrier integrity and hippocampal-dependent memory function, suggesting that complement activation may play a role in the mechanisms underlying PND development. Thus, interfering with the complement cascade, including C3, may provide promising therapeutic avenues for PND treatment. Indeed, aging and dementia, two well-established risk factors for delirium, are characterized by C3 accumulation on synapses. C3 gene deficiency reduced both synaptic loss near amyloid plaques and neurodegenerative pathology in an AD mouse model46. The promising protective effects of complement system inhibition were also described in a stroke mouse model. Administration of B4Crry, a selective complement inhibitor, prevents microglial phagocytosis of stressed neurons, thereby improving neuroinflammation and functional outcomes47. In addition to plasma C3, a study in hip fracture patients showed that the preoperative C3 concentration in cerebrospinal fluid (CSF) was significantly altered from normal levels in patients with postoperative delirium. Although the findings in two cohorts of patients were opposing, which may be due to multiple factors, including the duration of fracture and the sample analysis methodology48, the association between preoperative C3 in CSF and postoperative delirium should be noted and requires further validation.
Coagulation cascade after surgery
Complement signaling closely interacts with the coagulation cascade, which plays a critical role in the inflammatory response. Together with the release of DAMPs from the injury site, coagulation and thrombosis are activated after trauma and are critically implicated in restoring tissue homeostasis49. The fibrinolytic system, which leads to the conversion of fibrinogen to fibrin to initiate and resolve blood clotting, has well-established immune consequences. If fibrinogen enters the CNS parenchyma through a BBB opening, the insoluble fibrin becomes a strong immunogenic factor, known to bind to the CD11b I-domain of the CD11b/CD18 integrin receptor (also known as complement receptor 3 (CR3)) to further activate macrophages/resident microglia and drive cognitive deficits50,51,52. Notably, the CR3 can also recognize cleaved C3b (iC3b) and cause complement-dependent macrophage53 and microglia54 activation. We found perivascular fibrinogen deposition in the hippocampus as early as 24 hours after orthopedic surgery52,55,56. Therefore, fibrinogen may also represent a valuable peripheral biomarker to identify patients at risk for PNDs. Fibrin deposition has been implicated in several neurologic disorders, including multiple sclerosis, traumatic brain injury and AD (reviewed in ref. 57). Fibrin is also a well-established pathologic hallmark of BBB disruption and could represent a key mechanism for systemic inflammation leading to CNS inflammation and neuronal dysfunction after surgery. For example, plasma concentrations of the serine protease inhibitor PAI-1 are significantly associated with prolonged delirium in the emergency department and can be rapidly assayed in the circulation58. Indeed, peripheral biomarkers of inflammation and CNS dysfunction are becoming valuable tools for characterizing patient recovery59 and further highlight the complex interactions between systemic inflammation and CNS pathology. Thus, plasma biomarkers, including coagulation factors, lipid mediators and more classical proinflammatory cytokines, may already provide valuable information on the likelihood of developing PNDs without needing to directly access the CSF.
Blood–brain barrier opening and cell infiltration after surgery
BBB dysfunction is becoming more appreciated in the context of aging and dementia, although not many studies have characterized it in PNDs. One study evaluated changes in the pre- to postoperative serum albumin ratio in patients who had hip fracture surgery. Significant changes were measured in patients who developed delirium, suggesting that BBB dysfunction is relevant to the occurrence of postoperative cognitive complications60. Systemic biomarkers of BBB dysfunction and endothelial activation have also been described in critically ill patients with delirium61; thus, understanding the impact of inflammation on barrier function is paramount.
Preclinical models of postoperative neuroinflammation and PNDs have demonstrated that anesthesia and surgery can reduce tight junction (TJ) protein expression in the brain microvasculature, leading to increased BBB permeability and migration of CCR2+ ‘inflammatory’ macrophages into the hippocampus52,62,63. A simple laparotomy under isoflurane anesthesia causes cognitive impairment and increases BBB permeability in aged mice in an IL-6-dependent manner64. This change was accompanied by a decrease in β-catenin and TJ proteins such as claudin, occludin and zonula occudens-1 (ZO-1)64. Notably, in this study, exposure to anesthesia alone was not sufficient to trigger BBB opening even in aged mice. Using our orthopedic model, we also found no significant effects of well-balanced anesthesia alone on the CNS, including no inflammatory changes or behavioral deficits. A recent study in older adults that volunteered to receive anesthesia alone (without surgery) found no evidence of immune or neuronal injury as detected by plasma biomarkers65, suggesting that its combination with ensuing surgical trauma contributes to CNS impairments. After orthopedic surgery, blocking IL-6 receptor signaling with the monoclonal antibody tocilizumab effectively prevented BBB opening and the ensuing infiltration of CCR2+ cells into the hippocampus, and these effects were not observed in anesthesia-exposed mice66. Using the same tibial fracture surgery model in aged mice, T cell activation and IL-17A were implicated in BBB breakdown and PND behavior67. In this study, treatment with a monoclonal antibody reduced IL-17A expression in both the circulation and hippocampal tissue and also prevented BBB opening. Activation and degranulation of brain mast cells has also been causally implicated in microglial activation via MAPK signaling, resulting in neuronal loss in rats after orthopedic surgery68. Thus, multiple cell types, including neutrophils, BMDMs, mast and T cells, have been shown to be involved in PND pathology and its resolution. The mechanisms responsible for cell infiltration into the brain after peripheral surgery need further clarification.
Matrix-metalloproteinase (MMP) 2 and MMP9 are known regulators of TJs in brain capillaries and the BBB69. Peripheral surgery can elevate MMP2 and MMP9 expression in the hippocampus of aged mice and rats67,70. These changes are accompanied by a reduction in occludin and ZO-1. The role of MMP9 in surgery-induced BBB dysfunction and cognitive decline has been further validated using gene-targeted MMP9-deficient mice71.
The role of BBB disruption has been highlighted during aging and AD. A recent study described age-dependent BBB opening in mice by albumin extravasation in the hippocampus, starting as early as 12 months old72. The extravasated albumin is primarily taken up by astrocytes and contributes to neural dysfunction via transforming growth factor-β (TGF-β) signaling72. Notably, fibrinogen is a carrier of latent TGF-β, which induces astrogliosis and inhibits neurite outgrowth73. Indeed, disruption of the BBB is linked to multiple cell types involved in maintaining the neurovascular unit and may contribute to the onset of cognitive disorders. Age-dependent BBB breakdown has been described in humans using advanced, dynamic contrast-enhanced magnetic resonance imaging (MRI). In fact, progressive loss of hippocampal BBB integrity has already been measured in individuals without evident cognitive impairments74. This pathologic change is worsened in individuals with mild cognitive impairment and is associated with pericyte injury, as evidenced by an increase in the soluble growth factor receptor sPDGFR-β in CSF74. Taken together, during aging and neurodegeneration, peripheral inflammation can synergize with ongoing pathology, thus leading to worse outcomes in more frail individuals.
Glial activation after peripheral trauma
Microglia, the resident immune cells of the CNS, surveil the brain parenchyma and are rapidly activated following injury. Microglial ontogeny and biology have been extensively reviewed (for example, in ref. 75). Indeed, microglia and monocytes share the expression of several PRRs and can thus induce similar inflammatory responses. Microglia are central players in neuroinflammation, including inflammaging and neurodegeneration, and are one of the key cell types affected by surgical trauma. Their activation has been described in the human brain using positron emission tomography imaging, and it is associated with long lasting PNDs via higher [11C]PBR28 in patients with cognitive deficits after abdominal surgery76. PBR28 is a second-generation selective radiolabeled ligand for the 18-kDa translocator protein (TSPO), previously known as the peripheral benzodiazepine receptor, which is a widely expressed transmembrane protein that resides in the outer mitochondrial membrane of microglia but is also expressed in monocyte-derived macrophages77. Thus, changes in TSPO may not only reflect microglial dysfunction but also astrocytes and vascular endothelial cells, as well as systemic cells78,79. In this context, peripheral factors can activate microglia via a permeable BBB. In fact, mice treated with PLX5622, a selective inhibitor of the receptor CSF1R that can deplete microglia, and subjected to surgery remain protected from monocyte infiltration and do not develop PND behavior80. PLX5622 treatment is frequently billed as a highly specific method for microglial depletion81. Recent evidence indicates that it has minor yet consistent effects on other immune populations, namely, Ly6Clo ‘patrolling’ monocytes, which are reduced by as much as 30% with PLX5622 treatment80,82. Ablation of BMDMs using clodronate also prevents microglial activation after orthopedic surgery without altering monocyte chemoattractant protein-1 (MCP-1) expression in the brain. This suggests a critical role for resident microglia in actively recruiting peripheral leukocytes after surgery via MCP-1 signaling. Notably, increasing systemic concentrations of MCP-1 correlate with cognitive aging in older adults83. Nevertheless, the mechanisms that drive blood–CSF–brain communication need significant clarification in this context. MCP-1, together with a plethora of other chemokines, is upregulated in the systemic circulation after surgery, raising the possibility that other cellular sources, such as endothelial cells, can contribute to its upregulation in the CNS after surgery independently of BMDMs84.
With several factors able to affect microglia after surgery, a common feature described across PND models is the modification of cellular morphology, concomitant with the increased expression of molecules such as Iba-1 and CD11b on these cells. The effect of these changes on microglial function remains poorly defined. Recently, higher concentrations of the soluble receptor sTREM2 in CSF were detected in patients with delirium after hip fracture repair, confirming that microglial ‘activation’ occurs in the human brain due to ligands more specific than ubiquitous cytokines and chemokines85. TREM2 is a critical innate immune receptor that signals via the adaptor protein TYROBP/DAP12 and is expressed on microglia. Furthermore, TREM2 has established mutations associated with aging and neurodegenerative pathology75. Importantly, its signaling is essential to maintaining microglial homeostasis and overall metabolic fitness. In fact, dysregulation of TREM2 is a key functional signature of the disease-associated microglial profile86. During aging, TREM2 also contributes to microglial priming and inflammaging. Interestingly, several proinflammatory and oxidative genes, including C1q, C3 and CD11b, are downregulated in aged TREM2-deficient mice87. Whether TREM2 is causally related to PNDs is not yet known, and whether postoperative microglia express a disease-associated microglial profile is the focus of current studies using next-generation genetic sequencing approaches. From a histologic perspective, both microglia and astrocytes show distinct morphologic changes after peripheral trauma, and it is possible that proinflammatory factors released by microglia contribute to the subsequent activation of astrocytes, including IL-1α, TNF, and C1q88. Together, these mediators are both necessary and sufficient to activate A1 astrocytes, a more toxic and harmful subtype of these cells that are able to exert a potent neurotoxic effect. A1 astrocytes have been described in the context of normal aging, which may account for the higher risk of developing neurodegenerative conditions such as AD. Astrocytes are well known to shape synapses during development via complement signaling89. Notably, the aging brain expresses high levels of complement components, such as C1q, thus similar pathways may be engaged during aging as a result of microglial priming and stressors like lipopolysaccharide or surgery90,91,92. In this regard, A1 astrocytes may be responsible for the acute changes in neuronal plasticity and PND behavior, and studies are urgently needed to clarify these glial interactions (Fig. 2).

The proinflammatory surgical milieu (including cytokines, chemokines, DAMPs, coagulation factors, immunocytes, and so on) impacts the BBB, thus contributing to ensuing CNS pathology. Microglia, as first responders to changes in the CNS microenvironment, sense the changes following endothelial and BBB openings. Orthopedic surgery causes complement signaling activation in the brain and the deposition of fibrin(ogen) in the hippocampal brain parenchyma. Fibrin(ogen) and complement factors (such as activated forms of C3) are potent activators of microglia via CD11b signaling. Reactive microglia release proinflammatory factors, which further contribute to neuroinflammation. In particular, microglia can activate A1 astrocytes via IL-1, C1q and TNF to impact synapses and overall neuronal plasticity. Notably, the increased C3 in A1 astrocytes may in turn contribute to the microglial activation via C3aR. Surgery may also contribute to a DAM-like state, which requires further characterization in various preclinical models. Notably, sTREM2 is elevated in the CSF of patients with delirium after hip fracture, suggesting surgery impacts homeostatic DAM genes and microglial activity following surgical trauma.
Indeed, astrocytes are also key components of the BBB and closely interact with the cerebrovascular endothelium to maintain homeostasis. Surgery impairs expression of water channel proteins, such as aquaporin-4, which are critical to support astrocytic endfeet and BBB integrity55,56. The effect of astrocytic dysfunction after surgery is measurable in fluid biomarkers of postoperative patients, suggesting that CNS dysfunction is a result of peripheral trauma. For example, concentrations of the calcium-binding protein S100β and glial fibrillary acidic protein are elevated in the CSF and plasma of patients with cognitive impairments after cardiac and non-cardiac procedures93,94,95,96. Interestingly, for patients with pre-existing dementia, there is an even stronger association with these biomarkers, further demonstrating the higher vulnerability of this population to complications from these common procedures96. From these studies, it remains challenging to ascertain causality between different cell types and factors released in the brain parenchyma, especially with regard to astrocytic dysfunction that possibly precedes microgliosis. A recent study demonstrated that microglia migrate toward cerebral blood vessels after systemic inflammation97. Interestingly, the initial contact between microglia and cerebral vessels aims at preserving BBB integrity. However, with prolonged inflammation, microglia start to develop an activated phenotype and phagocytose astrocytic endfeet, thus leading to BBB breakdown97. Indeed, the signals that trigger the initial microglial migration require further investigation and, in the acute postoperative setting, may indicate a protective response by microglia in PNDs. Therefore, trauma can trigger both specific and localized CNS responses, and studies aimed at characterizing the peripheral and central ‘signalome’ after surgery may reveal unique pathways as well as potential PND therapeutic targets.
Potential therapies and concluding remarks
As reviewed here, the immune response to surgical trauma involves multiple compartments and, overall, is triggered to protect organs and restore homeostasis. Targeting inflammation in this context presents unique challenges as well as opportunities. Challenges include the heightened risk for immunosuppression and disrupted healing, a major concern for patients after surgery and a significant limitation of many biologics and immune blockers discussed here. Opportunities include the very acute time course of PND (as compared, for example, to other neurodegenerative disorders that manifest over several years) and the predictability of the outcome of a good portion of surgical cases in the context of elective procedures. This latter point is attractive because knowing the time of injury can ensure timely treatment and possibly prevent the CNS sequelae.
In line with these concepts, a couple of new therapeutic approaches are emerging in the field that desperately need attention. Resolution of inflammation is gaining considerable interest due to the safety profile of endogenous mediators and their potent actions on immune cells98. Indeed, it is now appreciated that the process of resolution is activated as soon as trauma occurs. This process is highly regulated by the biosynthesis of specialized proresolving lipid mediators (SPMs) from omega-3 polyunsaturated fatty acids, which include resolvins, protectins and maresins99. SPMs act as agonists to shorten the resolution interval of acute inflammation, affecting the influx and clearance of polymorphonuclear neutrophils already at the site of injury100,101. Importantly, they are not immunosuppressive, and, in the context of infection, SPMs can lower antibiotic requirements for bacterial clearance102. We have described the protective effects of D-series resolvins, including RvD1 and MaR1, which prevent surgery-induced microgliosis and PND-like behavior62,103. Importantly, SPM profiles determined by targeted metabololipidomics may serve as valuable biomarkers for CNS pathology. For example, patients with multiple sclerosis have altered SPM profiles in the blood that contribute to the regulation of monocytic migration through the BBB via pro-resolvin receptors expressed on endothelial cells104.We have found that surgery dynamically regulates expression of MaR1, an SPM specific to macrophages, in the human CSF, and this may have implications for BBB–monocyte transendothelial migration62.
SPMs are not the only target for PNDs, and other therapies aimed, for example, at stabilizing the BBB may precisely disengage this systemic-to-central immune response. One example is the use of targeted nanocarriers to finely regulate the cerebral vasculature, for example, via vascular cell adhesion molecule 1 (VCAM-1) expression105. Notably, endothelial VCAM-1 has been described as a key regulator of inflammation during aging, and administration of an anti-VCAM1 antibody reduces neuroinflammation and improves cognition in these mice106. We found that a mixed-lineage kinase (MLK) 3 inhibitor selectively protects the BBB after surgery without significantly affecting the systemic immune response or causing healing impairments at the fracture site following tibial fracture repair107. Finally, specific antibodies, such as 5B8, which targets the cryptic fibrin epitope γ377–395, thereby inhibiting fibrin-induced inflammation and oxidative stress without interfering with clotting108, may offer attractive strategies to safely prevent memory impairments. To date, only a few studies have evaluated the effects of anti-inflammatories on PNDs, partly because inflammation is a necessary response to injury, and tampering with this process can negatively impact healing and the host response. Thus, therapies that target dysfunctional inflammation may offer safer and more focused approaches to protect the brain from overactivation of the immune system.
In the Bucoliche (Egloga IX, written circa 39 bc), Virgil wrote “Time carries away everything, even our memory” (Omnia fert aetas, animum quoque). In this dialogue, Lycidas and Moeris reminisce on how difficult it has become to remember songs they used to sing in their youth and to recollect not only the actual words but also their intonation as they aged. This observation may be trivial. After all, we all have a hard time remembering things from the distant past. However, fast forward to the present time and, while forgetting remains a key manifestation of aging, it has also become an earlier pathologic hallmark of many debilitating neurologic disorders, including PNDs. If time slowly steals our memories, let us at least ensure that interventions like surgery do not accelerate the natural course of time, especially on an already vulnerable brain.
References
Pluvinage, J. V. & Wyss-Coray, T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat. Rev. Neurosci. 21, 93–102 (2020).
Soares, H. D., Hicks, R. R., Smith, D. & McIntosh, T. K. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 15, 8223–8233 (1995).
Gelderblom, M. et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857 (2009).
Evered, L. & Eckenhoff, R. G. & International Perioperative Cognition Nomenclature Working Group. Perioperative cognitive disorders. Response to: postoperative delirium portends descent to dementia. Br. J. Anaesth. 119, 1241 (2017).
Evered, L. A. & Silbert, B. S. Postoperative cognitive dysfunction and noncardiac surgery. Anesth. Analg. 127, 496–505 (2018).
Rasmussen, L. S. et al. Does anaesthesia cause postoperative cognitive dysfunction? A randomised study of regional versus general anaesthesia in 438 elderly patients. Acta Anaesthesiol. Scand. 47, 260–266 (2003).
Inouye, S. K., Westendorp, R. G. J. & Saczynski, J. S. Delirium in elderly people. Lancet 383, 911–922 (2014).
Koster, S., Hensens, A. G., Schuurmans, M. J. & van der Palen, J. Consequences of delirium after cardiac operations. Ann. Thorac. Surg. 93, 705–711 (2012).
Robinson, T. N. et al. Postoperative delirium in the elderly: risk factors and outcomes. Ann. Surg. 249, 173–178 (2009).
Marcantonio, E. R., Flacker, J. M., Michaels, M. & Resnick, N. M. Delirium is independently associated with poor functional recovery after hip fracture. J. Am. Geriatr. Soc. 48, 618–624 (2000).
Inouye, S. K. et al. The short-term and long-term relationship between delirium and cognitive trajectory in older surgical patients. Alzheimers Dement. 12, 766–775 (2016).
Saczynski, J. S. et al. Cognitive trajectories after postoperative delirium. N. Engl. J. Med. 367, 30–39 (2012).
Davis, D. H. J. et al. Association of delirium with cognitive decline in late life: a neuropathologic study of 3 population-based cohort studies. JAMA Psychiatry 74, 244–251 (2017).
Fong, T. G. et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology 72, 1570–1575 (2009).
Prince, M. et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63–75.e2 (2013).
Fick, D. M., Agostini, J. V. & Inouye, S. K. Delirium superimposed on dementia: a systematic review. J. Am. Geriatr. Soc. 50, 1723–1732 (2002).
Lee, H. B., Oldham, M. A., Sieber, F. E. & Oh, E. S. Impact of delirium after hip fracture surgery on one-year mortality in patients with or without dementia: a case of effect modification. Am. J. Geriatr. Psychiatry 25, 308–315 (2017).
AGS/NIA Delirium Conference Writing Group, Planning Committee and Faculty. The American Geriatrics Society/National Institute on Aging Bedside-to-Bench Conference: research agenda on delirium in older adults. J. Am. Geriatr. Soc. 63, 843–852 (2015).
Kotfis, K. et al. COVID-19: ICU delirium management during SARS-CoV-2 pandemic. Crit. Care 24, 176 (2020).
Leslie, D. L., Marcantonio, E. R., Zhang, Y., Leo-Summers, L. & Inouye, S. K. One-year health care costs associated with delirium in the elderly. Arch. Intern. Med. 168, 27–32 (2008).
Fong, T. G., Davis, D., Growdon, M. E., Albuquerque, A. & Inouye, S. K. The interface between delirium and dementia in elderly adults. Lancet Neurol. 14, 823–832 (2015).
Louveau, A., Harris, T. H. & Kipnis, J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577 (2015).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Zindel, J. & Kubes, P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu. Rev. Pathol. 15, 493–518 (2020).
Huber-Lang, M., Lambris, J. D. & Ward, P. A. Innate immune responses to trauma. Nat. Immunol. 19, 327–341 (2018).
Lotze, M. T. & Tracey, K. J. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342 (2005).
Scaffidi, P., Misteli, T. & Bianchi, M. E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 (2002).
Terrando, N. et al. Tumor necrosis factor-α triggers a cytokine cascade yielding postoperative cognitive decline. Proc. Natl Acad. Sci. USA 107, 20518–20522 (2010).
Vacas, S., Degos, V., Tracey, K. J. & Maze, M. High-mobility group box 1 protein initiates postoperative cognitive decline by engaging bone marrow–derived macrophages. Anesthesiology 120, 1160–1167 (2014).
Venereau, E. et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209, 1519–1528 (2012).
Lu, S.-M. et al. S100A8 contributes to postoperative cognitive dysfunction in mice undergoing tibial fracture surgery by activating the TLR4/MyD88 pathway. Brain Behav. Immun. 44, 221–234 (2015).
Fonken, L. K. et al. The alarmin HMGB1 mediates age-induced neuroinflammatory priming. J. Neurosci. 36, 7946–7956 (2016).
Terrando, N. et al. Systemic HMGB1 neutralization prevents postoperative neurocognitive dysfunction in aged rats. Front. Immunol. 7, 441 (2016).
Lamkanfi, M. et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185, 4385–4392 (2010).
Cape, E. et al. Cerebrospinal fluid markers of neuroinflammation in delirium: a role for interleukin-1β in delirium after hip fracture. J. Psychosom. Res. 77, 219–225 (2014).
Cibelli, M. et al. Role of interleukin-1β in postoperative cognitive dysfunction. Ann. Neurol. 68, 360–368 (2010).
Zheng, B., Lai, R., Li, J. & Zuo, Z. Critical role of P2X7 receptors in the neuroinflammation and cognitive dysfunction after surgery. Brain Behav. Immun. 61, 365–374 (2017).
Ji, S.-R., Wu, Y., Potempa, L. A., Liang, Y.-H. & Zhao, J. Effect of modified C-reactive protein on complement activation: a possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 26, 935–941 (2006).
Sjöberg, A. P., Trouw, L. A., McGrath, F. D. G., Hack, C. E. & Blom, A. M. Regulation of complement activation by C-reactive protein: targeting of the inhibitory activity of C4b-binding protein. J. Immunol. 176, 7612–7620 (2006).
Bíro, A. et al. Studies on the interactions between C-reactive protein and complement proteins. Immunology 121, 40–50 (2007).
Suresh, M. V., Singh, S. K., Ferguson, D. A. Jr. & Agrawal, A. Role of the property of C-reactive protein to activate the classical pathway of complement in protecting mice from pneumococcal infection. J. Immunol. 176, 4369–4374 (2006).
Hecke, F. et al. Circulating complement proteins in multiple trauma patients—correlation with injury severity, development of sepsis, and outcome. Crit. Care Med. 25, 2015–2024 (1997).
Thordardottir, S., Vikingsdottir, T., Bjarnadottir, H., Jonsson, H. Jr. & Gudbjornsson, B. Activation of complement following total hip replacement. Scand. J. Immunol. 83, 219–224 (2016).
Hoedemaekers, C. et al. The complement system is activated in a biphasic pattern after coronary artery bypass grafting. Ann. Thorac. Surg. 89, 710–716 (2010).
Xiong, C. et al. Complement activation contributes to perioperative neurocognitive disorders in mice. J. Neuroinflammation 15, 254 (2018).
Shi, Q. et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9, eaaf6295 (2017).
Alawieh, A., Langley, E. F. & Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 10, eaao6459 (2018).
Westhoff, D. et al. Preoperative protein profiles in cerebrospinal fluid in elderly hip fracture patients at risk for delirium: a proteomics and validation study. BBA Clin. 4, 115–122 (2015).
Reis, E. S., Mastellos, D. C., Hajishengallis, G. & Lambris, J. D. New insights into the immune functions of complement. Nat. Rev. Immunol. 19, 503–516 (2019).
Adams, R. A. et al. The fibrin-derived γ377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J. Exp. Med. 204, 571–582 (2007).
Merlini, M. et al. Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron 101, 1099–1108.e6 (2019).
Terrando, N. et al. Resolving postoperative neuroinflammation and cognitive decline. Ann. Neurol. 70, 986–995 (2011).
Takizawa, F., Tsuji, S. & Nagasawa, S. Enhancement of macrophage phagocytosis upon iC3b deposition on apoptotic cells. FEBS Lett. 397, 269–272 (1996).
Ramaglia, V. et al. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl Acad. Sci. USA 109, 965–970 (2012).
Velagapudi, R. et al. Orthopedic surgery triggers attention deficits in a delirium-like mouse model. Front. Immunol. 10, 2675 (2019).
Wang, P. et al. Neurovascular and immune mechanisms that regulate postoperative delirium superimposed on dementia. Alzheimers Dement. 16, 734–749 (2020).
Petersen, M. A., Ryu, J. K. & Akassoglou, K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat. Rev. Neurosci. 19, 283–301 (2018).
McNeil, J. B. et al. Plasma biomarkers of inflammation, coagulation, and brain injury as predictors of delirium duration in older hospitalized patients. PLoS ONE 14, e0226412 (2019).
Khan, B. A. et al. Biomarkers of delirium duration and delirium severity in the ICU. Crit. Care Med. 48, 353–361 (2020).
Hov, K. R. et al. Blood-cerebrospinal fluid barrier integrity in delirium determined by Q-albumin. Dement. Geriatr. Cogn. Disord. 41, 192–198 (2016).
Hughes, C. G. et al. Endothelial activation and blood-brain barrier injury as risk factors for delirium in critically ill patients. Crit. Care Med. 44, e809–e817 (2016).
Yang, T. et al. Maresin 1 attenuates neuroinflammation in a mouse model of perioperative neurocognitive disorders. Br. J. Anaesth. 122, 350–360 (2019).
Degos, V. et al. Depletion of bone marrow–derived macrophages perturbs the innate immune response to surgery and reduces postoperative memory dysfunction. Anesthesiology 118, 527–536 (2013).
Yang, S. et al. Anesthesia and surgery impair blood–brain barrier and cognitive function in mice. Front. Immunol. 8, 902 (2017).
Deiner, S. et al. Human plasma biomarker responses to inhalational general anaesthesia without surgery. Br. J. Anaesth. 125, 282–290 (2020).
Hu, J. et al. Interleukin-6 is both necessary and sufficient to produce perioperative neurocognitive disorder in mice. Br. J. Anaesth. 120, 537–545 (2018).
Ni, P. et al. IL-17A contributes to perioperative neurocognitive disorders through blood-brain barrier disruption in aged mice. J. Neuroinflammation 15, 332 (2018).
Zhang, X. et al. Activated brain mast cells contribute to postoperative cognitive dysfunction by evoking microglia activation and neuronal apoptosis. J. Neuroinflammation 13, 127 (2016).
Rempe, R. G. et al. Matrix metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J. Neurosci. 38, 4301–4315 (2018).
Li, Z. et al. Surgery-induced hippocampal angiotensin II elevation causes blood-brain barrier disruption via MMP/TIMP in aged rats. Front. Cell Neurosci. 10, 105 (2016).
Bi, J., Shan, W., Luo, A. & Zuo, Z. Critical role of matrix metallopeptidase 9 in postoperative cognitive dysfunction and age-dependent cognitive decline. Oncotarget 8, 51817–51829 (2017).
Senatorov, V. V. Jr. et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 11, eaaw8283 (2019).
Schachtrup, C. et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 30, 5843–5854 (2010).
Montagne, A. et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015).
Song, W. M. & Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 19, 1048–1058 (2018).
Forsberg, A. et al. The immune response of the human brain to abdominal surgery. Ann. Neurol. 81, 572–582 (2017).
Owen, D. R. et al. Determination of [11C]PBR28 binding potential in vivo: a first human TSPO blocking study. J. Cereb. Blood Flow Metab. 34, 989–994 (2014).
Notter, T. et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol. Psychiatry 23, 323–334 (2018).
Cosenza-Nashat, M. et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 35, 306–328 (2009).
Feng, X. et al. Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI Insight 2, e91229 (2017).
Huang, Y. et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 21, 530–540 (2018).
Valdearcos, M. et al. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab. 26, 185–197.e3 (2017).
Bettcher, B. M. et al. Increases in a pro-inflammatory chemokine, MCP-1, are related to decreases in memory over time. Front. Aging Neurosci. 11, 25 (2019).
Deshmane, S. L., Kremlev, S., Amini, S. & Sawaya, B. E. Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29, 313–326 (2009).
Henjum, K. et al. CSF sTREM2 in delirium—relation to Alzheimer’s disease CSF biomarkers Aβ42, t-tau and p-tau. J. Neuroinflammation 15, 304 (2018).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Linnartz-Gerlach, B. et al. TREM2 triggers microglial density and age-related neuronal loss. Glia 67, 539–550 (2019).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Stephan, A. H. et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474 (2013).
Hennessy, E., Griffin, É. W. & Cunningham, C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J. Neurosci. 35, 8411–8422 (2015).
Clarke, L. E. et al. Normal aging induces A1-like astrocyte reactivity. Proc. Natl Acad. Sci. USA 115, E1896–E1905 (2018).
Rappold, T. et al. Evidence of an association between brain cellular injury and cognitive decline after non-cardiac surgery. Br. J. Anaesth. 116, 83–89 (2016).
Lopez, M. G. et al. Intraoperative oxidative damage and delirium after cardiac surgery. Anesthesiology 132, 551–561 (2020).
Hall, R. J. et al. Delirium and cerebrospinal fluid S100B in hip fracture patients: a preliminary study. Am. J. Geriatr. Psychiatry 21, 1239–1243 (2013).
Hov, K. R. et al. Cerebrospinal fluid S100B and Alzheimer’s disease biomarkers in hip fracture patients with delirium. Dement. Geriatr. Cogn. Dis. Extra 7, 374–385 (2017).
Haruwaka, K. et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 10, 5816 (2019).
Serhan, C. N. & Levy, B. D. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 128, 2657–2669 (2018).
Serhan, C. N. & Savill, J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 (2005).
Bannenberg, G. L. et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 (2005).
Malawista, S. E., de Boisfleury Chevance, A., van Damme, J. & Serhan, C. N. Tonic inhibition of chemotaxis in human plasma. Proc. Natl Acad. Sci. USA 105, 17949–17954 (2008).
Chiang, N. et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528 (2012).
Terrando, N. et al. Aspirin-triggered resolvin D1 prevents surgery-induced cognitive decline. FASEB J. 27, 3564–3571 (2013).
Kooij, G. et al. Specialized pro-resolving lipid mediators are differentially altered in peripheral blood of patients with multiple sclerosis and attenuate monocyte and blood-brain barrier dysfunction. Haematologica 105, 2056–2070 (2020).
Marcos-Contreras, O. A. et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl Acad. Sci. USA 117, 3405–3414 (2020).
Yousef, H. et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 25, 988–1000 (2019).
Miller-Rhodes, P. et al. The broad spectrum mixed-lineage kinase 3 inhibitor URMC-099 prevents acute microgliosis and cognitive decline in a mouse model of perioperative neurocognitive disorders. J. Neuroinflammation 16, 193 (2019).
Ryu, J. K. et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 19, 1212–1223 (2018).
Acknowledgements
We thank Kathy Gage (Duke University Medical Center) for editorial assistance. T.Y. acknowledges a Department of Medicine Chair Research Award (Duke University) and an ASN Kidney Cure Carl W. Gottschalk Research Scholar Grant. N.T. is supported by NIA grants R01AG057525, R21 AG055877-01A1 and R03 AG064260-01; the Department of Anesthesiology; and Duke Institute of Brain Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information L. A. Dempsey was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, T., Velagapudi, R. & Terrando, N. Neuroinflammation after surgery: from mechanisms to therapeutic targets. Nat Immunol 21, 1319–1326 (2020). https://doi.org/10.1038/s41590-020-00812-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-020-00812-1
This article is cited by
-
Itaconate alleviates anesthesia/surgery-induced cognitive impairment by activating a Nrf2-dependent anti-neuroinflammation and neurogenesis via gut-brain axis
Journal of Neuroinflammation (2024)
-
Complement C3 From Astrocytes Plays Significant Roles in Sustained Activation of Microglia and Cognitive Dysfunctions Triggered by Systemic Inflammation After Laparotomy in Adult Male Mice
Journal of Neuroimmune Pharmacology (2024)
-
Mind the Gap: Unraveling the Intricate Dance Between Alzheimer’s Disease and Related Dementias and Bone Health
Current Osteoporosis Reports (2024)
-
Tregs dysfunction aggravates postoperative cognitive impairment in aged mice
Journal of Neuroinflammation (2023)
-
Risk factors for delayed neurocognitive recovery in elderly patients undergoing thoracic surgery
BMC Anesthesiology (2023)
