
Abstract
Allium crop breeding remains severely hindered due to the lack of high-quality reference genomes. Here we report high-quality chromosome-level genome assemblies for three key Allium crops (Welsh onion, garlic and onion), which are 11.17 Gb, 15.52 Gb and 15.78 Gb in size with the highest recorded contig N50 of 507.27 Mb, 109.82 Mb and 81.66 Mb, respectively. Beyond revealing the genome evolutionary process of Allium species, our pathogen infection experiments and comparative metabolomic and genomic analyses showed that genes encoding enzymes involved in the metabolic pathway of Allium-specific flavor compounds may have evolved from an ancient uncharacterized plant defense system widely existing in many plant lineages but extensively boosted in alliums. Using in situ hybridization and spatial RNA sequencing, we obtained an overview of cell-type categorization and gene expression changes associated with spongy mesophyll cell expansion during onion bulb formation, thus indicating the functional roles of bulb formation genes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
The raw sequencing data of onion, garlic, Welsh onion and Africa lily were deposited in the National Center for Biotechnology Information Sequence Read Archive under the accession PRJNA948806 and in the National Genomics Data Center (https://ngdc.cncb.ac.cn/?lang=en) under the accession PRJCA016760. The assemblies of the four genomes reported in this paper have been deposited in the National Center for Biotechnology Information under accessions JASDDO000000000, JASFAV000000000, JASFAW000000000, JASFAX000000000 and in the Genome Warehouse in National Genomics Data Center128,129, Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under accession GWHCBHY00000000, GWHCBHZ00000000, GWHCBIA00000000 and GWHCBIB00000000 that are publicly accessible at https://ngdc.cncb.ac.cn/gwh.
Code availability
All software used in the study are publicly available on the Internet as described in the Methods and Reporting Summary.
Change history
14 December 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41588-023-01610-9
References
Chase, M. W. et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016).
Jones, M. G. et al. Biosynthesis of the flavour precursors of onion and garlic. J. Exp. Bot. 55, 1903–1918 (2004).
Yoshimoto, N. & Saito, K. S-Alk (en)ylcysteine sulfoxides in the genus Allium: proposed biosynthesis, chemical conversion, and bioactivities. J. Exp. Bot. 70, 4123–4137 (2019).
Reis, A. C. et al. rDNA mapping, heterochromatin characterization and AT/GC content of Agapanthus africanus (L.) Hoffmanns (Agapanthaceae). An. Acad. Bras. Cienc. 88, 1727–1734 (2016).
Sharaibi, O. J. & Afolayan, A. J. Micromorphological characterization of the leaf and rhizome of Agapanthus praecox subsp. praecox Willd. (Amaryllidaceae). J. Bot. 2017, 1–10 (2017).
Fenwick, G. R., Hanley, A. B. & Whitaker, J. R. The genus Allium—part 1. Crit. Rev. Food Sci. Nutr. 22, 199–271 (1985).
Boulos, L. Flora of Egypt (Al Hadara Publishing, 1999).
Harris, J., Cottrell, S., Plummer, S. & Lloyd, D. Antimicrobial properties of Allium sativum (garlic). Appl. Microbiol. Biotechnol. 57, 282–286 (2001).
Capasso, A. Antioxidant action and therapeutic efficacy of Allium sativum L. Molecules 18, 690–700 (2013).
Borlinghaus, J., Albrecht, F., Gruhlke, M. C., Nwachukwu, I. D. & Slusarenko, A. J. Allicin: chemistry and biological properties. Molecules 19, 12591–12618 (2014).
Fu, J. et al. Identification and characterization of abundant repetitive sequences in Allium cepa. Sci. Rep. 9, 16756 (2019).
Khandagale, K. et al. Omics approaches in Allium research: progress and way ahead. PeerJ 8, e9824 (2020).
King, J., Bradeen, J., Bark, O., McCallum, J. & Havey, M. J. A low-density genetic map of onion reveals a role for tandem duplication in the evolution of an extremely large diploid genome. Theor. Appl. Genet. 96, 52–62 (1998).
Jakše, J. et al. Pilot sequencing of onion genomic DNA reveals fragments of transposable elements, low gene densities, and significant gene enrichment after methyl filtration. Mol. Genet. Genomics 280, 287–292 (2008).
Shigyo, M., Khar, A. & Abdelrahman, M. (eds) The Allium Genomes pp. 99–112 (Springer, 2018).
Kiseleva, A., Kirov, I. & Khrustaleva, L. Chromosomal organization of centromeric Ty3/gypsy retrotransposons in Allium cepa L. and Allium fistulosum L. Russ. J. Genet. 50, 586–592 (2014).
Hertweck, K. L. Assembly and comparative analysis of transposable elements from low coverage genomic sequence data in Asparagales. Genome 56, 487–494 (2013).
Peška, V., Mandáková, T., Ihradská, V. & Fajkus, J. Comparative dissection of three giant genomes: Allium cepa, Allium sativum, and Allium ursinum. Int. J. Mol. Sci. 20, 733 (2019).
Sun, X. et al. A chromosome-level genome assembly of garlic (Allium sativum) provides insights into genome evolution and allicin biosynthesis. Mol. Plant 13, 1328–1339 (2020).
Ohri, D., Fritsch, R. M. & Hanelt, P. Evolution of genome size in Allium (Alliaceae). Plant Syst. Evol. 210, 57–86 (1998).
Duchoslav, M., Šafářová, L. & Jandová, M. Role of adaptive and non-adaptive mechanisms forming complex patterns of genome size variation in six cytotypes of polyploid Allium oleraceum (Amaryllidaceae) on a continental scale. Ann. Bot. 111, 419–431 (2013).
Khrustaleva, L., Kudryavtseva, N., Romanov, D., Ermolaev, A. & Kirov, I. Comparative tyramide-FISH mapping of the genes controlling flavor and bulb color in Allium species revealed an altered gene order. Sci. Rep. 9, 12007 (2019).
Shigyo, M., Khar, A. & Abdelrahman, M. (eds) The Allium Genomes pp 197–214 (Springer, 2018).
Ricroch, A., Yockteng, R., Brown, S. C. & Nadot, S. Evolution of genome size across some cultivated Allium species. Genome 48, 511–520 (2005).
Liao, N. et al. Chromosome-level genome assembly of bunching onion illuminates genome evolution and flavor formation in Allium crops. Nat. Commun. 13, 6690 (2022).
Finkers, R. et al. Insights from the first genome assembly of onion (Allium cepa). G3 (Bethesda) 11, jkab243 (2021).
Nystedt, B. et al. The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584 (2013).
Neale, D. B. et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 15, R59 (2014).
Guan, R. et al. Draft genome of the living fossil Ginkgo biloba. GigaScience 5, 49 (2016).
Li, G. et al. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat. Genet. 53, 574–584 (2021).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245 (2019).
Ming, R. et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 47, 1435–1442 (2015).
Harkess, A. et al. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 8, 1279 (2017).
Zhang, Y. et al. Chromosome-scale assembly of the Dendrobium chrysotoxum genome enhances the understanding of orchid evolution. Hortic. Res. 8, 183 (2021).
Magallón, S., Gómez‐Acevedo, S., Sánchez‐Reyes, L. L. & Hernández‐Hernández, T. A metacalibrated time‐tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207, 437–453 (2015).
Zhang, G. Q. et al. The Apostasia genome and the evolution of orchids. Nature 549, 379–383 (2017).
Wang, X. et al. Genome alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Mol. Plant 8, 885–898 (2015).
Sensalari, C., Maere, S. & Lohaus, R. ksrates: positioning whole-genome duplications relative to speciation events in KS distributions. Bioinformatics 38, 530–532 (2022).
Yamaguchi, Y. & Kumagai, H. Characteristics, biosynthesis, decomposition, metabolism and functions of the garlic odour precursor, S‑allyl‑L‑cysteine sulfoxide. Exp. Ther. Med. 19, 1528–1535 (2020).
Michelmore, R. W. & Meyers, B. C. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 8, 1113–1130 (1998).
Guo, L. et al. The opium poppy genome and morphinan production. Science 362, 343–347 (2018).
Yuan, M. et al. Pattern-recognition receptors are required for NLR-mediated plant immunity. Nature 592, 105–109 (2021).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327 (2002).
Chen, A. et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. Preprint at bioRxiv https://doi.org/10.1101/2021.01.17.427004 (2021).
Xia, K. et al. The single-cell stereo-seq reveals region-specific cell subtypes and transcriptome profiling in Arabidopsis leaves. Dev. Cell 57, 1299–1310 (2022).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792 (2022).
Zhang, C. et al.Transcriptome sequencing and metabolism analysis reveals the role of cyanidin metabolism in dark-red onion (Allium cepa L.) bulbs. Sci. Rep. 8, 14109 (2018).
Goławska, S., Sprawka, I., Łukasik, I. & Goławski, A. Are naringenin and quercetin useful chemicals in pest-management strategies? J. Pest Sci. 87, 173–180 (2014).
Kurepa, J., Shull, T. E. & Smalle, J. A. Quercetin feeding protects plants against oxidative stress (version 1; peer review: 1 approved, 1 approved with reservations). F1000Res. 5, 2430 (2016).
Sossountzov, L. et al. Spatial and temporal expression of a maize lipid transfer protein gene. Plant Cell 3, 923–933 (1991).
Suh, M. C. et al. Cuticular lipid composition, surface structure, and gene expression in Arabidopsis stem epidermis. Plant Physiol. 139, 1649–1665 (2005).
DeBono, A. et al. Arabidopsis LTPG is a glycosylphosphatidylinositol-anchored lipid transfer protein required for export of lipids to the plant surface. Plant Cell 21, 1230–1238 (2009).
Yeats, T. H. & Rose, J. K. The biochemistry and biology of extracellular plant lipid‐transfer proteins (LTPs). Protein Sci. 17, 191–198 (2008).
Heath, O. Formative effects of environmental factors as exemplified in the development of the onion plant. Nature 155, 623–626 (1945).
Mita, T. & Shibaoka, H. Changes in microtubules in onion leaf sheath cells during bulb development. Plant Cell Physiol. 24, 109–117 (1983).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Qiu, X. et al. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315 (2017).
Zhang, C. et al. Transcriptome analysis of sucrose metabolism during bulb swelling and development in onion (Allium cepa L.). Front. Plant Sci. 7, 1425 (2016).
Atif, M. J. et al. Mechanism of Allium crops bulb enlargement in response to photoperiod: a review. Int. J. Mol. Sci. 21, 1325 (2020).
Shibaoka, H. Plant hormone-induced changes in the orientation of cortical microtubules: alterations in the cross-linking between microtubules and the plasma membrane. Annu. Rev. Plant Biol. 45, 527–544 (1994).
Zhong, R., Burk, D. H., Morrison, W. H. & Ye, Z. H. A kinesin-like protein is essential for oriented deposition of cellulose microfibrils and cell wall strength. Plant Cell 14, 3101–3117 (2002).
Ganguly, A., Zhu, C., Chen, W. & Dixit, R. FRA1 kinesin modulates the lateral stability of cortical microtubules through cellulose synthase–microtubule uncoupling proteins. Plant Cell 32, 2508–2524 (2020).
Rao, G., Zeng, Y., He, C. & Zhang, J. Characterization and putative post-translational regulation of α-and β-tubulin gene families in Salix arbutifolia. Sci. Rep. 6, 19258 (2016).
Goley, E. D. & Welch, M. D. The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 7, 713–726 (2006).
Singh, R. et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 500, 335–339 (2013).
Paterson, A. H., Bowers, J. E. & Chapman, B. A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl Acad. Sci. USA 101, 9903–9908 (2004).
Tuskan, G. A. et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604 (2006).
Vanneste, K., Baele, G., Maere, S. & Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Res. 24, 1334–1347 (2014).
D'hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217 (2012).
Putnik, P. et al. An overview of organosulfur compounds from Allium spp.: from processing and preservation to evaluation of their bioavailability, antimicrobial, and anti-inflammatory properties. Food Chem. 276, 680–691 (2019).
Ellmore, G. S. & Feldberg, R. S. Alliin lyase localization in bundle sheaths of the garlic clove (Allium sativum). Am. J. Bot. 81, 89–94 (1994).
Stotz, H. U. et al. Role of camalexin, indole glucosinolates, and side chain modification of glucosinolate‐derived isothiocyanates in defense of Arabidopsis against Sclerotinia sclerotiorum. Plant J. 67, 81–93 (2011).
Hématy, K. et al. Moonlighting function of phytochelatin synthase1 in extracellular defense against fungal pathogens. Plant Physiol. 182, 1920–1932 (2020).
Matern, A. et al. A substrate of the ABC transporter PEN3 stimulates bacterial flagellin (flg22)-induced callose deposition in Arabidopsis thaliana. J. Biol. Chem. 294, 6857–6870 (2019).
Birnbaum, K. et al. A gene expression map of the Arabidopsis root. Science 302, 1956–1960 (2003).
Jean-Baptiste, K. et al. Dynamics of gene expression in single root cells of Arabidopsis thaliana. Plant Cell 31, 993–1011 (2019).
Shulse, C. N. et al. High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep. 27, 2241–2247 (2019).
Olsen, J. L. et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335 (2016).
Shusei, S. et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Project, A. G. et al. The Amborella genome and the evolution of flowering plants. Science 342, 1241089 (2013).
Hyde, P. T., Earle, E. D. & Mutschler, M. A. Doubled haploid onion (Allium cepa L.) lines and their impact on hybrid performance. HortScience 47, 1690–1695 (2012).
Healey, A., Furtado, A., Cooper, T. & Henry, R. J. Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 10, 21 (2014).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Xie, T. et al. De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana. Mol. Plant 8, 489–492 (2015).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with Hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 1–27 (2020).
Fajkus, P. et al. Allium telomeres unmasked: the unusual telomeric sequence (CTCGGTTATGGG) n is synthesized by telomerase. Plant J. 85, 337–347 (2016).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Tichenor, C. A new software metric to complement function points: the software non-functional assessment process (SNAP). https://apps.dtic.mil/sti/pdfs/ADA592012.pdf (2013).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 10, 547–548 (2000).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 15–30 (2018).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Bell, C. D., Soltis, D. E. & Soltis, P. S. The age and diversification of the angiosperms re‐revisited. Am. J. Bot. 97, 1296–1303 (2010).
Xu, Y. et al. Corrigendum #2 to ‘VGSC: a web-based vector graph toolkit of genome synteny and collinearity’. BioMed Res. Int. 2019, 2150291 (2019).
Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986).
Sun, P. et al. WGDI: a user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 15, 1841–1851 (2022).
Soltis, P. S. & Soltis, D. E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 30, 159–165 (2016).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Sayyari, E., Whitfield, J. B. & Mirarab, S. DiscoVista: interpretable visualizations of gene tree discordance. Mol. Phylogenet. Evol. 122, 110–115 (2018).
Zwaenepoel, A. & Van de Peer, Y. wgd—simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 35, 2153–2155 (2019).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 1–14 (2009).
Sato, K., Tanaka, T., Shigenobu, S., Motoi, Y. & Itoh, T. Improvement of barley genome annotations by deciphering the Haruna Nijo genome. DNA Res. 23, 21–28 (2016).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Wang, H. et al. Identification of antibiotic resistance genes in the multidrug-resistant Acinetobacter baumannii strain, MDR-SHH02, using whole-genome sequencing. Int. J. Mol. Med. 39, 364–372 (2017).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A. & Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121 (2013).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Carmona-Saez, P., Chagoyen, M., Tirado, F., Carazo, J. & Pascual-Montano, A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 8, R3 (2007).
Huang, T. et al. Deciphering the effects of gene deletion on yeast longevity using network and machine learning approaches. Biochimie 94, 1017–1025 (2012).
Chen, L., Li, B.-Q. & Feng, K.-Y. Predicting biological functions of protein complexes using graphic and functional features. Curr. Bioinform. 8, 545–551 (2013).
Chen, M. et al. Genome warehouse: a public repository housing genome-scale data. Genom. Proteom. Bioinform. 19, 584–589 (2021).
CNCB-NGDC Members and PartnersDatabase resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 51, D18–D28 (2023).
Acknowledgements
We thank M. J. Havey from the Department of Horticulture, University of Wisconsin (Madison, WI, USA) for providing the onion seeds of the doubled haploid line and L. Cui from the Liaoning Academy of Agricultural Sciences (Shenyang, Liaoning, China) for providing the inbred lines of Welsh onion. We thank Y. Gu, L. Chen, Q. Lin, L. Chen, B. Mu, H. Sun, X. Wei, J. Li, S. Li, H. Lu and S. Zhang for general technical assistance or discussion. We thank G. Zhang from Zhejiang University and K. Wang from Northwestern Polytechnical University (NWPU) for their comments and suggestions when preparing our manuscript. We thank Y. Zeng from BGI-Shenzhen for sharing his idea on Allium genome study. This work was supported by the Thousand Talents Plan (5113190037), the Talents Team Construction Fund of NWPU and the Fundamental Research Funds for the Central Universities (3102019JC007) to J.C.; the Talents Team Construction Fund of NWPU and the Projects of Interdisciplinary of NWPU (0202022GH0306) to W.W.; National Natural Science Foundation of China (21801206), the Joint Research Funds of Department of Science & Technology of Shaanxi Province and NWPU (2020GXLH-Z-015) to Z.R.; Guangdong Provincial Key Laboratory of Genome Read and Write (2017B030301011) to X.X.
Author information
Authors and Affiliations
Contributions
F.H. and J.C. managed the project. J.C., W.W., R.M., H.Y. and X.X. conceived the study. F.H. and J.C. wrote the manuscript with contributions from all other authors. B.Z. and H.Z. were responsible for genome assembly into contigs and Hi-C scaffolding. X.L. handled genome annotation and analysis of phylogeny, transcriptome, gene family evolution, allicin/isoallicin pathway and immune system. B.Z., Z.T., X.L. and Z.L. were responsible for the analysis of whole-genome synteny and whole-genome duplication (WGD). Z.T. and P.Z. handled repeat annotation and conducted analysis of LTR retrotransposons. L.Z., J.H., J.Q., Q.L., Y.Z. and K.W. were responsible for in situ hybridization experiments. L.Z. focused on studying the evolution and formation of tunicated bulbs. H.Z. was responsible for genome evaluation and conducted ultra-high-performance liquid chromatography/tandem mass spectrometry (UHPLC-MS/MS) analysis. H.Z., K.X., X.G., L.L., W.S., B.Z., S.L. and L.P. conducted spatial RNA sequencing and analyzed spatial transcriptome data. Y.P., W.Z., F.L., Z.R. and J.M. were in charge of plant material collection and DNA/RNA preparation. H.Y., L.H. and W.C. supervised genome sequencing and library construction and also collected plant material for bulb development.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Zhangjun Fei and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Genome overview and phylogenetic analyses of Allium species and African lily, related to Fig. 1.
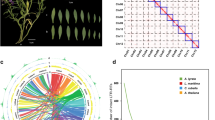
a, Overview of genomes of Allium species and African lily: track a corresponds to chromosome length. From b to d, three rings represent density distribution of different genomic features, including coding gene, GC, and repeat sequences, respectively. Track e corresponds to syntenic blocks. b, Species tree by summarizing each gene tree with Astral. c, Phylogenies from three supergene matrice, each of which was constructed by concatenating ortholog gene sets defined by reciprocal best hit (RBH) by BLAST, single-copy genes (SCG) identified using OrthoFinder and single-copy genes (SCG) identified using OrthoMCL, respectively. d, Single-copy genes identified using OrthoMCL. e, Single-copy genes identified using OrthoFinder. f, Relative evolutionary rate comparison among three Allium species and African lily.
Extended Data Fig. 2 Analysis of repetitive sequences in three Allium species and African lily.
a, Overall composition of repetitive elements in different genomes. DNA, DNA element; LINE, long interspersed nuclear element; LTR, long terminal repeat transposable element; SINE, short interspersed nuclear element. Divergence distribution of Copia (b) and Gypsy (c) retrotransposons in four assembled genomes. X-axis represents divergence measured in percentage of sequence differences with consensus in TE library. d, Phylogenetic relationships of Gypsy and Copia retrotransposon domains across genomes of asparagus, rice, and four species sequenced in this study (Supplementary Data 3 for alignments of sequences and a version with support values on branches).
Extended Data Fig. 3 Evolution of gene families, related to Fig. 3.
a, Pie diagram on each branch of tree represents the proportion of gene families undergoing gain (green) or loss (red) events. Numbers below pie diagram denote total number of expanded and contracted gene families. b, Distribution of single-copy, multiple-copy, unique, and other orthologs in 10 plant species, and three columns on right show number of genes in families, family number, and average genes per family in 10 plant species. c, Point-line and Venn diagrams represent shared and unique gene families among 10 species or four closely related Amaryllidaceae species (Welsh onion, garlic, onion, and African lily). Each number represents number of gene families. d, Significant GO terms of molecular function, biological process, and cellular component enriched in expanded gene families in ancestral branch of three Allium plants. e, Significant KEGG pathways enriched in expanded gene families in ancestral branch of three Allium plants. f, Network diagram of significant KEGG pathways enriched in expanded gene families in ancestral branch of three Allium plants. GO terms or KEGG pathways probably related to alliinase, plant immune system, and bulb development discussed in this study are labeled with red arrows. The p values (unadjusted, one side) presented in d and e were calculated using the hypergeometric test.
Extended Data Fig. 4 Clustering of alliinase gene family in Allium and outgroup species, related to Fig. 3.
Phylogenetic analysis of alliinase gene family, with alliinase genes forming 15 distinct groups (A–O). Bootstrap values are shown on each branch. For the alliinase gene family, we constructed a gene tree using standard maximum-likelihood phylogenetic analysis implemented in IQ-TREE with default parameters and 1,000 bootstraps.
Extended Data Fig. 5 Clustering of lachrymatory factor synthase gene family (LFS) in Allium, related to Fig. 3.
Phylogenetic analysis of LFS gene family, with LFS genes forming eight distinct groups (A–H). Bootstrap values are shown on each branch. For LFS gene family, we constructed a gene tree using standard maximum-likelihood phylogenetic analysis implemented in IQ-TREE with default parameters and 1,000 bootstraps.
Extended Data Fig. 6 Location of genes associated with allicin/isoallicin biosynthesis on chromosomes.
Localization of genes related to allicin/isoallicin biosynthetic pathways on chromosomes of a, Welsh onion, b, garlic, and c, onion.
Extended Data Fig. 7 Gene expression changes upon pathogen infection in onion.
a, Relative expression levels of alliin biosynthesis genes in transcriptome of fungal-infected blades. b, Significant KEGG pathways enriched in up-regulated genes in response to fungal infection. Comparing gene expression profiles between leaf blades of fungal-infected and healthy plants, we identified 1,035 up-regulated genes during infection. c, Relative expression levels of alliin biosynthesis genes after artificial injury and exposure to bacterial culture by qRT-PCR. d and e, Relative expression levels of homologous genes of alliin biosynthesis genes in Arabidopsis thaliana. TPM: transcripts per kilobase of exon model per million mapped reads. Transcriptome data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142747. Four-week-old Arabidopsis thaliana plant leaves were infiltrated with sterile water (Mock) or different Pst strains, then harvested at 3 or 6 h after infiltration. Two bacterial strains, P. s. pv. tomato (Pst) DC3000 (avrRpt2), which activates RPS2 (resistance to P. syringae 2)-dependent effector-triggered immunity (ETI) and pattern-triggered immunity (PTI), and Pst DC3000 without ‘avirulent’ effector to activate PTI only in wild-type plants. Data presented in a, c–e are mean ± standard error of three or four independent experiments, and bars with p values were significantly different based on t test (two-sided). The p values (unadjusted, one side) presented in b were calculated using the hypergeometric test.
Extended Data Fig. 8 Cell marker genes in different clusters, related to Fig. 4.
a, Dot plot showing expression profiles of marker genes in all 14 cell clusters. ID of cell marker genes in onion are below x-axis. Expression level for each bin was calculated by scaled number of molecular identifiers (MIDs) for each marker gene. Average expression (AE) level of all bins in each cell cluster is denoted by dot color. Percentage of bins expressing marker gene (PE) in each cell cluster is denoted by dot size. b, Spatial expression patterns of cell marker genes from in situ hybridization (left) and spatial RNA-seq (right), showing that in situ hybridization and stereo-seq data were highly consistent and supported cell categorization. Scale bars equal to 1 mm for in situ hybridization, 800 um for spatial RNA-seq. Number in parenthesis represents number of samples with same signal pattern over total number of samples.
Extended Data Fig. 9 Spatial expression patterns of genes related to synthesis of flavonoid compounds and cuticular wax, related to Fig. 4e,f.
a, Spatial expression patterns of genes (CHS, CHIL, CHI, F3H, F3'H, LTP2 and LTP3) in different onion bulb development stages based on stereo-seq data. Co-expression of genes in epidermal cells was observed in all samples, showing that flavonoid and cuticular wax biosynthesis is mainly active in epidermal cells. b, Expression pattern of LTP in leaf base of onion by RNA in situ hybridization. Lower panels are images with reverse colors produced by ImageJ based on images in upper panels. Expression patterns based on in situ hybridization confirmed LTP results in Fig. 4f from large-scale Stereo-seq analysis. Scale bars equal to 800 μm for a, 1 mm for b. Number in parenthesis represents number of samples with same signal pattern over total number of samples.
Extended Data Fig. 10 Spongy mesophyll cell lineages and onion bulb formation, related to Figs. 4g,5.
a, Visualization of spongy mesophyll cell lineages, including six sections at three development stages. Sections from different stages and individuals show similar patterns of spongy mesophyll cell development: spongy mesophyll cells from inner to outer layer, and for each layer from base to top and from outer to inner, represent early to late points along the expansion process. Progression of numbers from small to large indicates pseudotime sequence, reflecting progression of spongy mesophyll cell expansion. Same color pattern is also used in panel b. b and c, UMAP dimensionality reduction projection of spongy mesophyll cells grouped by pseudotime scores and sections, respectively. General pattern of pseudotime increasing from left to right can be seen in panel b, but no clear pattern can be seen in c, suggesting that expansion of spongy mesophyll cell development is a fundamentally short process, starting asynchronously at different development stages as indicated in panel a. d and e, Spatial visualization of expression of indicated genes related to onion bulb formation. Scale bars equal to 800 μm for a, d and e.
Supplementary information
Supplementary Information
Supplementary Results 1–6, Supplementary Methods and Supplementary Figs. 1–20.
Supplementary Tables
Supplementary Tables 1–54.
Supplementary Data 1
Sequence alignment for the analysis of positive selection of genes in the three Allium species.
Supplementary Data 2
Sequence alignment of alliinase gene family and LFS gene family.
Supplementary Data 3
Sequence alignment of phylogenetic relationships of Gypsy and Copia retrotransposon domains across genomes of asparagus, rice and four species sequenced in this study, and a version with support values on branches.
Supplementary Data 4
Sequence alignment of positively selected or rapidly evolving genes involved in bulb formation in onion or garlic.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hao, F., Liu, X., Zhou, B. et al. Chromosome-level genomes of three key Allium crops and their trait evolution. Nat Genet 55, 1976–1986 (2023). https://doi.org/10.1038/s41588-023-01546-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-023-01546-0
