
Abstract
IgA nephropathy (IgAN) is a progressive form of kidney disease defined by glomerular deposition of IgA. Here we performed a genome-wide association study of 10,146 kidney-biopsy-diagnosed IgAN cases and 28,751 controls across 17 international cohorts. We defined 30 genome-wide significant risk loci explaining 11% of disease risk. A total of 16 loci were new, including TNFSF4/TNFSF18, REL, CD28, PF4V1, LY86, LYN, ANXA3, TNFSF8/TNFSF15, REEP3, ZMIZ1, OVOL1/RELA, ETS1, IGH, IRF8, TNFRSF13B and FCAR. The risk loci were enriched in gene orthologs causing abnormal IgA levels when genetically manipulated in mice. We also observed a positive genetic correlation between IgAN and serum IgA levels. High polygenic score for IgAN was associated with earlier onset of kidney failure. In a comprehensive functional annotation analysis of candidate causal genes, we observed convergence of biological candidates on a common set of inflammatory signaling pathways and cytokine ligand–receptor pairs, prioritizing potential new drug targets.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

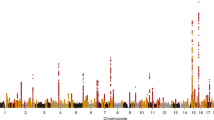
Data availability
Primary genotype data for previously published cohorts are available through dbGAP under accession number phs000431.v2.p1, and the new genotype data are available under dbGAP accession number phs000431.v3.p1. Our IRB determined that the use of the primary genotype data is restricted to genetic studies of kidney disease. GWAS summary statistics are available from the Kiryluk Lab website: https://www.columbiamedicine.org/divisions/kiryluk/study_gwas_stat_IgA_summary.php. The PAGE consortium control genotype data are available on dbGAP under accession number phs000356.v2.p1. The eMERGE-III imputed genotype and phenotype data are available through dbGAP, accession number phs001584.v2.p2. The UKBB genotype and phenotype data are available through the UKBB web portal https://www.ukbiobank.ac.uk/. The DICE dataset is available at https://dice-database.org/; the Blood eQTL meta-analysis by eQTLGen is available at https://www.eqtlgen.org/; the Kidney eQTL Atlas is available at http://susztaklab.com/eqtl; NEPTUNE eQTL Browser is available at http://nephqtl.org/; GTEx is available at https://gtexportal.org/home/; GWAS catalog is available at https://www.ebi.ac.uk/gwas; LD Hub is available at http://www.nealelab.is/tools-and-software; Open Targets platform is available at https://www.targetvalidation.org; DLRP can be accessed at https://dip.doe-mbi.ucla.edu/dip/DLRP.cgi; DrugBank can be accessed at https://www.drugbank.ca; GlobalData database is available at https://www.globaldata.com/industries-we-cover/pharmaceutical; FUN-LDA scores are available at http://www.columbia.edu/~ii2135/funlda.html; MSigDB and GSEA are available at http://software.broadinstitute.org/gsea/msigdb/; STRING is available at https://string-db.org; InWeb is available at http://www.lagelab.org/resources/; the Metabolomics GWAS Server is available at http://metabolomics.helmholtz-muenchen.de/gwas/; and the MGI database is available at http://www.informatics.jax.org.
Code availability
Only publicly available open-source software was used in the analyses; there was no custom software.
References
Feehally, J. et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J. Am. Soc. Nephrol. 21, 1791–1797 (2010).
Gharavi, A. G. et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 43, 321–327 (2011).
Kiryluk, K. et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 8, e1002765 (2012).
Kiryluk, K. et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 46, 1187–1196 (2014).
Yu, X. Q. et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 44, 178–182 (2012).
Li, M. et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat. Commun. 6, 7270 (2015).
Li, M. et al. Genome-wide meta-analysis identifies three novel susceptibility loci and reveals ethnic heterogeneity of genetic susceptibility for IgA nephropathy. J. Am. Soc. Nephrol. 31, 2949–2963 (2020).
Kiryluk, K., Novak, J. & Gharavi, A. G. Pathogenesis of immunoglobulin A nephropathy: recent insight from genetic studies. Annu Rev. Med. 64, 339–356 (2013).
Kiryluk, K. & Novak, J. The genetics and immunobiology of IgA nephropathy. J. Clin. Invest. 124, 2325–2332 (2014).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Feriozzi, S. & Polci, R. The role of tonsillectomy in IgA nephropathy. J. Nephrol. 29, 13–19 (2016).
Liu, L. et al. Genetic regulation of serum IgA levels and susceptibility to common immune, infectious, kidney, and cardio-metabolic traits. Nat. Commun. 13, 6859 (2022).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Backenroth, D. et al. FUN-LDA: a latent dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation: methods and applications. Am. J. Hum. Genet. 102, 920–942 (2018).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
Iotchkova, V. et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat. Genet. 51, 343–353 (2019).
Harley, J. B. et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 50, 699–707 (2018).
Zhou, H. et al. Epstein–Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 17, 205–216 (2015).
Sugai, M., Watanabe, K., Nambu, Y., Hayashi, T. & Shimizu, A. Functions of Runx in IgA class switch recombination. J. Cell. Biochem. 112, 409–414 (2011).
Malhotra, N. & Kang, J. SMAD regulatory networks construct a balanced immune system. Immunology 139, 1–10 (2013).
Lage, K. et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 25, 309–316 (2007).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220 (2010).
Han, S. S. et al. The role of TNF superfamily member 13 in the progression of IgA nephropathy. J. Am. Soc. Nephrol. 27, 3430–3439 (2016).
Yamada, K. et al. Leukemia inhibitory factor signaling enhances production of galactose-deficient IgA1 in IgA nephropathy. Kidney Dis. (Basel) 6, 168–180 (2020).
Suzuki, H. et al. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J. Biol. Chem. 289, 5330–5339 (2014).
Yamada, K. et al. Inhibition of STAT3 signaling reduces IgA1 autoantigen production in IgA nephropathy. Kidney Int. Rep. 2, 1194–1207 (2017).
Vosa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Owen, C. R., Yuan, L. & Basson, M. D. Smad3 knockout mice exhibit impaired intestinal mucosal healing. Lab. Invest. 88, 1101–1109 (2008).
Yang, X. et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 18, 1280–1291 (1999).
Agarwal, S. et al. Human Fc receptor-like 3 inhibits regulatory T cell function and binds secretory IgA. Cell Rep. 30, 1292–1299 (2020).
Tolnay, M. Lymphocytes sense antibodies through human FCRL proteins: emerging roles in mucosal immunity. J. Leukoc. Biol. 111, 477–487 (2021).
Choobdar, S. et al. Assessment of network module identification across complex diseases. Nat. Methods 16, 843–852 (2019).
Zhang, X. et al. An integrative investigation of the therapeutic mechanism of Ainsliaea fragrans Champ. in cervicitis using liquid chromatography tandem mass spectrometry based on a rat plasma metabolomics strategy. J. Pharm. Biomed. Anal. 156, 221–231 (2018).
Zhao, L. et al. Stachydrine ameliorates isoproterenol-induced cardiac hypertrophy and fibrosis by suppressing inflammation and oxidative stress through inhibiting NF-kappaB and JAK/STAT signaling pathways in rats. Int. Immunopharmacol. 48, 102–109 (2017).
Fulco, C. P. et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, bax028 (2017).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021).
Zipfel, P. F. et al. Complement inhibitors in clinical trials for glomerular diseases. Front. Immunol. 10, 2166 (2019).
Titze, S. et al. Disease burden and risk profile in referred patients with moderate chronic kidney disease: composition of the German Chronic Kidney Disease (GCKD) cohort. Nephrol. Dial. Transpl. 30, 441–451 (2015).
Wunnenburger, S. et al. Associations between genetic risk variants for kidney diseases and kidney disease etiology. Sci. Rep. 7, 13944 (2017).
Hansen, I. S., Baeten, D. L. P. & den Dunnen, J. The inflammatory function of human IgA. Cell. Mol. Life Sci. 76, 1041–1055 (2019).
Infante, B. et al. Recurrence of immunoglobulin A nephropathy after kidney transplantation: a narrative review of the incidence, risk factors, pathophysiology and management of immunosuppressive therapy. Clin. Kidney J. 13, 758–767 (2020).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Robertson, C. C. et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat. Genet. 53, 962–971 (2021).
Fang, H. et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 51, 1082–1091 (2019).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Shi, J. & Lee, S. A novel random effect model for GWAS meta-analysis and its application to trans-ethnic meta-analysis. Biometrics 72, 945–954 (2016).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 8, e64683 (2013).
Pillai, N. E. et al. Predicting HLA alleles from high-resolution SNP data in three Southeast Asian populations. Hum. Mol. Genet. 23, 4443–4451 (2014).
Ka, S. et al. HLAscan: genotyping of the HLA region using next-generation sequencing data. BMC Bioinformatics 18, 258 (2017).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Tian, C. et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599 (2017).
Vilhjalmsson, B. J. et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015).
Khera, A. V. et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177, 587–596 (2019).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Gorski, M. et al. 1000 Genomes-based meta-analysis identifies 10 novel loci for kidney function. Sci. Rep. 7, 45040 (2017).
Pattaro, C. et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun. 7, 10023 (2016).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Heng, T. S. & Painter, M. W. & Immunological Genome Project Consortium. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008).
Eppig, J. T. et al. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 43, D726–D736 (2015).
Bult, C. J. et al. Mouse genome database 2016. Nucleic Acids Res. 44, D840–D847 (2016).
Hormozdiari, F., Kostem, E., Kang, E. Y., Pasaniuc, B. & Eskin, E. Identifying causal variants at loci with multiple signals of association. Genetics 198, 497–508 (2014).
Schmiedel, B. J. et al. Impact of genetic polymorphisms on human immune. Cell 175, 1701–1715 (2018).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Qiu, C. et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 24, 1721–1731 (2018).
Suhre, K. et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 477, 54–60 (2011).
Shin, S. Y. et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 46, 543–550 (2014).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Suhre, K. et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 8, 14357 (2017).
Emilsson, V. et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 361, 769–773 (2018).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Li, T. et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat. Methods 14, 61–64 (2017).
Su, G., Kuchinsky, A., Morris, J. H., States, D. J. & Meng, F. GLay: community structure analysis of biological networks. Bioinformatics 26, 3135–3137 (2010).
Nepusz, T., Yu, H. & Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 9, 471–472 (2012).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 (2009).
Graeber, T. G. & Eisenberg, D. Bioinformatic identification of potential autocrine signaling loops in cancers from gene expression profiles. Nat. Genet. 29, 295–300 (2001).
Knox, C. et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 39, D1035–D1041 (2011).
Zhu, F. et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 40, D1128–D1136 (2012).
Carvalho-Silva, D. et al. Open targets platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065 (2019).
Ochoa, D. et al. Open targets platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. 49, D1302–D1310 (2021).
Levey, A. S. et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 150, 604–612 (2009).
Schwartz, G. J. et al. New equations to estimate GFR in children with CKD. J. Am. Soc. Nephrol. 20, 629–637 (2009).
Stanaway, I. B. et al. The eMERGE genotype set of 83,717 subjects imputed to ~40 million variants genome wide and association with the herpes zoster medical record phenotype. Genet. Epidemiol. 43, 63–81 (2019).
Khan, A. et al. Medical records-based genetic studies of the complement system. J. Am. Soc. Nephrol. 32, 2031–2047 (2021).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Denny, J. C. et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210 (2010).
Acknowledgements
We are grateful to all study participants across multiple nephrology centers worldwide for their contributions to this manuscript. This work was supported by the following institutions, grants and funding agencies: Columbia University, Columbia Glomerular Center, IGA Nephropathy Foundation of America and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01-DK105124 (to K.K., J. Novak, B.A.J.), RC2-DK116690 (to K.K.) and R01-DK082753 (to A.G.G., J. Novak, K.K., F.S., B.A.J., R.J.W.). Additional support was provided by R01LM013061 (to K.K.), U01HG008680 (to K.K.), U01AI152960 (to K.K.), R01-DK078244 (to J. Novak, B.A.J.), R01-AI149431 (to J. Novak, B.A.J.), National Science Foundation of China (82022010 to X.-J.Z.; 82070733 to H.Z.), Beijing Natural Science Foundation (Z190023 to X.-J.Z.), Deutsche Forschungsgemeinschaft (DFG) Project-ID 192904750—CRC 992 Medical Epigenetics (to A. Köttgen, P. Schlosser), DFG Project-ID 431984000—CRC 1453 (to A. Köttgen) and EQUIP Program for Medical Scientists, Faculty of Medicine, University of Freiburg (to P. Schlosser). J.B.H., S.P., L.K. and M.W. were supported by R01HG010730, U01AI130830, R01NS099068, R01AI024717, R01AR073228, U01AI150748, R01AI148276, P01AI150585 and the US Department of Veterans Affairs Merit Award I01-BX001834 to J.B.H. D.P.G. was supported by the St Peter’s Trust for Kidney, Bladder and Prostate Research. The UK cohort data were generated as a result of a grant from Kidney Research UK and the Medical Research Council (to J. Barratt and D.P.G.). The German STOP-IgAN study was supported by the DFG (German Research Foundation)—CRU 5011—Project-ID 445703531 (to J.F., T.R.). The GCKD study was funded by grants from the German Ministry of Education and Research (BMBF, 01ER0804) and the KfH Foundation for Preventive Medicine. Unregistered grants to support the study were provided by Bayer, Fresenius Medical Care and Amgen. Genotyping was supported by Bayer Pharma AG. The recruitment of Polish cases with IgAN was sponsored by the Polish Kidney Genetics Network (POLYGENES), a collaborative effort between Columbia University and Poznań University of Medical Sciences, Poland. The recruitment of Czech patients with IgAN was supported by the research project of General University Hospital in Prague (RVO-VFN64165). The recruitment of the Russian cohort was supported by the Government Assignment of the Russian Ministry of Health, Assignment 200080056 of the Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University. The recruitment of the Swedish cohort was supported by the Nephrology and Rheumatology Departments at Karolinska University Hospital and Karolinska Institutet, Stockholm, and grants from the Swedish Society of Medicine. We thank the Immunopathology Working Group of the Italian Society of Nephrology (ISN) for inviting their member sites to contribute to this study. We are also grateful to the Pediatric Nephrology Research Consortium (PNRC) for hosting and cosponsoring the GIGA-kids study that recruited pediatric patients with IgAN across the PNRC sites. We are additionally grateful to C. Bowers for coordinating GIGA-kids and PMRC-based recruitment, J. Narus for coordinating recruitment at the University of Utah, E. Elenberg and S. Shah for helping enroll participants from Texas Children’s Hospital and A. Suwanichkul for sample shipping. The recruitment of the Korean cohort was supported by the Seoul National University Hospital Human Biobank, a member of the National Biobank of Korea, financed by the Ministry of Health and Welfare, Republic of Korea. We are grateful to the International Multiple Sclerosis Genetics Consortium (IMSGC), the Inflammatory Bowel Disease Genetics Consortium (IBDGC), the International Myositis Consortium (IMC), the Feinstein Institute for Medical Research, the French Biological Ressource Center for MS Genetics (REFGENSEP), Genethon and INSERM for contributing Immunochip controls for the purpose of this study. We also thank the Population Architecture Using Genomics and Epidemiology (PAGE) consortium, funded by the NHGRI with cofunding from the NIMHD, for providing population controls genotyped with MEGA chip for this study. Molecular graphics shown in Extended Data Fig. 5c were generated using UCSF Chimera, developed by the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. The funding sources were not involved in the study design, collection, analysis and interpretation of data, writing of the report or the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
K.K. and A.G.G. conceived the study, provided overall supervision of the project and made the decision to publish the findings. E.S.-R. and N. Mladkova performed quality control, imputation and association analyses for GWAS and Immunochip discovery cohorts. E.S.-R. performed final statistical analyses, meta-analyses, fine-mapping studies, functional annotations and drug-target analyses. F.Z. and A. Khan designed polygenic risk scores, tested for genome-wide genetic correlations and performed clinical correlation analyses. L.L. performed analysis of the HLA locus. J.B.H., M.W., S.P. and L.K. performed RELI transcription factor analysis. I.I.-L. consulted on the statistics and functional annotation of GWAS loci. K.K., A.G.G. and J. Novak provided biological interpretation of GWAS loci. X.-J.Z. and H.Z. coordinated recruitment, genotyping and analysis of the Beijing cohorts. F.S. coordinated recruitment activities across the Italian network of clinical recruitment sites. Y.C. coordinated recruitment activities across the Turkish recruitment sites. H.T. coordinated recruitment across the Argentinian network of clinical recruitment sites. V.T. and D.M. coordinated the recruitment of the Czech cohort. J.X. and N.C. coordinated recruitment, genotyping and analysis of the Shanghai cohorts. H.S. recruited and clinically characterized the Japanese discovery cohort. H.L., J.-H.P., B.C., Y.S.K. and D.K.K. recruited and clinically characterized the Korean discovery cohort. J.F. and T.R. contributed DNA and clinical data for the German STOP-IgAN cohort. K.K., R.N., R.J.W. and W.S. led the GIGA-kids study in collaboration with the Pediatric Nephrology Research Consortium (PNRC). M. Marasa, O.B. and J.Y.Z. coordinated recruitment at Columbia University and managed clinical data and DNA samples. J. Barratt and D.P.G. provided summary statistics for the UK GWAS cohort. R.P.L. contributed previously published European and Beijing GWAS cohorts. A.B.E., K.-U.E. and A. Köttgen contributed German cases to the Immunochip discovery cohorts, and P. Schlosser performed polygenic risk score validation studies in the GCKD cohort. D.v.H., C.C., C.W., A.F., V.A. and P.K.G. contributed Illumina Immunochip idat files for population controls. R.J.F.L. and E.E.K. contributed Illumina MEGA chip idat files for population controls. S.S.-C., A.S.B., P.A.C., G.B.A., J.R., B. Sprangers, D.C.C., H.R., Y.P., P.R., K.G., B. Stengel, M. Metzger, G. Canaud, N. Maillard, F.B., L. Berthelot, E.P., R. Monteiro, J.M., A.-A.S., G.H., A.Q., P.W., R.S., D. Selewski, K. Davis, M. Kallash, T.L.V., M.R., A.C., D. Ranch, S.E.W., D. Samsonov, D.J.C., O.A., D.G., M. Stangou, J. Nagy, T.K., E.F., A.A., C.B., D.C., L.D.V., G.G.B., M.B., E.B., L. Bono, G.B., G. Caridi, F. Lugani, G. Ghiggeri, R.C., L. Peruzzi, V.E., C.E., S.F., R.P., G.F., M. Galliani, M. Garozzo, A. Mitrotti, L.G., S. Granata, G.Z., F. Londrino, R. Magistroni, I.P., A. Magnano, C.M., P.M., R. Mignani, A.P., C.P., D. Roccatello, M. Salvadori, E.S., D. Santoro, G. Gembillo, S.S., D. Spotti, P.Z., C.I., F.A., E.D., M.F., N. Krata, K.M., L. Pączek, S.N., B.M., M.P.-T., M.M.-W., A.P.-P., T.B., M.D., K.P., P.S., M.Z., D.K., M. Krajewska, I.K.-G., Z.H., B.B.-P., T.L., A.D.-S., T.H., A.M.-K., M. Miklaszewska, M. Szczepańska, K. Dyga, E.M., K.S.-L., M.P.-B., M.T., D. Runowski, N. Kwella, D.D., I.H., F.K., L. Prikhodina, B.F., I.N., S. Goto, M.C., A.V.W., A.H., E.A., J. Ballarin, S.L., B.V., L.-Y.M., T.A., P.A.K., U.P. and B.A.J. contributed to the recruitment and clinical characterization of patients with IgAN and controls recruited from their respective clinical centers. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.A.J. and J. Novak are cofounders, co-owners of, and consultants for Reliant Glycosciences, LLC, and are co-inventors on US patent application 14/318,082 (assigned to UAB Research Foundation). A.G.G. has served on an advisory board for Novartis, Travere and Natera and receives research grant funding from the Renal Research Institute and Natera. K.K. has served on an advisory board for Goldfinch Bio and Gilead. The other authors report no competing interests.
Peer review
Peer review information
Nature Genetics thanks Xueqing Yu, Richard Oram and Amy McKnight for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quantile-quantile (QQ) plot of the combined meta-analysis across 38,897 individuals.
Blue dots represent the QQ plot based on all SNPs in the meta-analysis, and red dots represent the QQ plot after exclusion of SNPs within the MHC region. The overall genomic inflation factor (λ) was 1.048 with the MHC region, and 1.042 without the MHC region. The y-axis depicts −log10 of the observed two-sided P-values for a fixed effects meta-analysis of all cohorts without correction for multiple testing. The x-axis depicts −log10 of the expected two-sided P-values under the null hypothesis.
Extended Data Fig. 2 Regional plots for non-HLA genome-wide significant loci.
a, The FCRL locus. b, The TNFSF4 locus. c, The CFH locus. d, The REL locus. e, The CD28 locus. f, The PF4V1/CXCL8 locus. g, The IRF4 locus. h, The RREB1 locus. i, The DEFA locus. j, The LYN locus. k, The ANXA3 locus. l, The TNFSF8 locus. m, The CARD9 locus. n, The REEP3 locus. o, The ZMIZ1 locus. p, The RELA locus. q, The ETS1 locus. r, The IGH locus. s, The ITGAM locus. t, The IRF8 locus. u, The TNFSF13 locus. v, The TNFRSF13B locus. w, The FCAR locus. x, The HORMAD2/LIF locus. The x-axis shows the physical position in Mb (hg19 coordinates) and known genes. The left y-axis presents −log10 of two-sided P-values for variant association statistics (fixed effects meta-analysis under an additive model without correction for multiple testing), and the right y-axis shows the recombination rate across the region. The dotted horizontal line indicates a genome-wide significance threshold of 5.0 × 10−8.
Extended Data Fig. 3 Manhattan plots for East Asian GWAS subgroup analyses.
a, East Asian meta-analysis under an additive genetic model (λ = 1.040) revealed a new genome-wide significant locus on chr. 6 (CCR6, green). b, East Asian meta-analysis under a recessive model (λ = 0.940) revealed a new genome-wide significant locus on chr. 1 (encoding PADI3 and PADI4, green). The y-axis shows -log10 of two-sided P-values for a fixed effects meta-analysis under an additive or recessive genetic model without correction for multiple testing (note that y-axis is truncated to accommodate the HLA peak). The x-axis shows genomic position along each chromosome (1-22 and X). The dotted horizontal line indicates P = 5.0 × 10−8.
Extended Data Fig. 4 Stepwise conditional analyses of the CFH and HLA loci.
a, CFH locus. Initial meta-analysis results without conditioning (top left); after conditioning for the top SNP rs6677604 (top middle); and after controlling for the two significant SNPs rs6677604 and rs12029571 (top right). b, HLA locus. Initial meta-analysis results without conditioning (middle left); after conditioning for the top SNP rs9268557 (middle), after controlling for rs9268557 and rs9275355 (middle right), after controlling for rs9268557, rs9275355 and rs9272105 (bottom left), after controlling for rs9268557, rs9275355, rs9272105 and rs9275596 (bottom middle), and after controlling for rs9268557, rs9275355rs9272105, rs9275596 and rs3128927 (bottom right) with no additional significant signals. The x-axis shows genomic position in Mb (hg19 coordinates) and known genes. The left y-axis presents −log10 P-values for association statistics (two-sided P-values for a fixed effects meta-analysis under an additive genetic model without correction for multiple testing). The right y-axis (light-blue line) shows the average recombination rate across the region. The dotted horizontal line indicates a genome-wide significance threshold of 5.0 × 10−8. The top SNP in each panel is marked by a red diamond.
Extended Data Fig. 5 Stepwise conditional analysis of imputed polymorphic amino-acid positions in DRβ, DQβ, and DQα peptides in East Asian and European cohorts.
a, East Asian cohorts. b, European cohorts. Each symbol represents a polymorphic site tested for association with IgAN along the peptide sequence. The x-axis shows genomic position of the sequence encoding each amino acid. The y-axis shows global statistical significance for each polymorphic site (two-sided multiallelic Wald test without adjustment for multiple testing). The dashed horizontal line corresponds to a genome-wide significance threshold of 5.0 × 10−8. c, Physical location of independently associated amino acid positions 11 (red) and 71 (orange) in the structural model of DRβ. The DR structure was visualized using UCSF Chimera v1.16 based on Protein Data Bank entry 3PDO.
Extended Data Fig. 6 Enrichment tests for the GWAS candidate gene set against human ortholog gene sets that when genetically manipulated in mice result in “Abnormal Antibody Levels” or “Abnormal Immunoglobulin A Levels”.
a, Plot for “Abnormal Antibody Levels”. b, Plot for “Abnormal Immunoglobulin A Levels”. The x-axis depicts gene rank by statistical significance. The y-axis depicts −log10 of two-sided P-value based on the enrichment test without multiple testing correction. The red horizontal line is the Bonferroni-corrected statistical significance level corrected for the number of genes tested.
Extended Data Fig. 7 Pathway, cell type and tissue enrichment analyses.
a,b, MAGMA pathway enrichment analysis based on GWAS summary statistics with (a) and without (b) the HLA region. −log10 of two-sided P-values for the enrichment test (not corrected for multiple testing) are depicted along the x-axis. Significant pathways are listed along the y-axis. Red vertical lines indicate significance threshold after accounting for multiple testing. c, Cell type-specific heritability enrichment for functional annotations in ImmGen dataset of mouse regulatory elements and expression data demonstrating the strongest enrichment in small intestine-derived macrophages profiled three days after Salmonella infection. d, Cell type-specific heritability enrichment for individual functional annotations generated by Roadmap Epigenomics demonstrates significant enrichment in immune cells, especially of B-cell lineage. All enrichments at nominal two-sided uncorrected P < 0.05 are displayed and grouped according to cell and tissue class. A solid red line represents a stringent Bonferroni-corrected −log10 of the two-sided P-value threshold of significance for the Roadmap dataset (P = 1.3 × 10−4). A dotted black line represents the −log10 of the nominal P-value of 0.05. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MF, macrophages; P/I, post-injection.
Extended Data Fig. 8 Cis-regulatory effects at the CARD9 locus.
a, The CARD9 gene model depicting 13 exons of CARD9 and the splicing events from GTEx blood RNA-seq. The top IgAN risk allele (rs4077515-T) encodes S12N substitution in the second exon of CARD9. This SNP also exhibits a strong and statistically significant blood sQTL effect, wherein the risk allele is associated with higher rates of the chr9:136372094:136373532:clu_47895 splicing event, leading to the retention of exon 2 (red) in the coding sequence of CARD9 (isoform A), while the protective (rs4077515-C) allele is associated with the alternative splicing event that truncates exon 2 (isoform B). The functional CARD domain (green) maps to a portion of exon 2 that is intact in isoform A, but truncated in isoform B. b, Blood splice QTL violin plots of normalized intron excision ratios (corresponding to the ratio of isoform A to isoform B) by the genotype of rs10870077 (top blood sQTL in GTEx) and rs4077515 (top SNP in GWAS for IgAN). These two SNPs are in near perfect linkage disequilibrium (r2 > 0.98). The white bar represents a median, the thick gray bar represents an interquartile range, and the blue shape reflects the distribution kernel density estimation. Numbers in parentheses under the x-axis indicate n independent samples per each genotype group. c, Both rs10870077 and rs4077515 are also associated with a significant cis-eQTL effect on CARD9 mRNA levels in GTEx blood (violin plots with similar definitions as in b). d, The blood eQTL signal for CARD9 significantly co-localizes with the GWAS signal (PP4 of 0.86). The top panel represents a regional plot for the GWAS signal (Immunochip data excluded). The bottom panel represents a blood cis-eQTL signal for CARD9 from the QTLGen Consortium meta-analysis (y-axis truncated at P < 1 × 10−310); rs10870077 and rs4077515 are indicated in red. All P-values are two-sided, correspond to a logistic regression Wald test under an additive genetic model, and are not corrected for multiple testing.
Supplementary information
Supplementary Information
Supplementary Tables 1–32, Note and References.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kiryluk, K., Sanchez-Rodriguez, E., Zhou, XJ. et al. Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy. Nat Genet 55, 1091–1105 (2023). https://doi.org/10.1038/s41588-023-01422-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-023-01422-x
This article is cited by
-
Use of corticosteroids in Norwegian patients with immunoglobulin a nephropathy progressing to end-stage kidney disease: a retrospective cohort study
BMC Nephrology (2024)
-
Genetics of IgA nephrology: risks, mechanisms, and therapeutic targets
Pediatric Nephrology (2024)
-
Sibeprenlimab bei Patienten mit IgA-Nephropathie
Die Nephrologie (2024)
-
Clinical Application of Polygenic Risk Score in IgA Nephropathy
Phenomics (2024)
-
IgA nephropathy
Nature Reviews Disease Primers (2023)
