
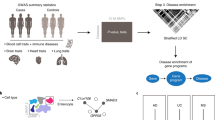
Abstract
Single-cell RNA sequencing (scRNA-seq) provides unique insights into the pathology and cellular origin of disease. We introduce single-cell disease relevance score (scDRS), an approach that links scRNA-seq with polygenic disease risk at single-cell resolution, independent of annotated cell types. scDRS identifies cells exhibiting excess expression across disease-associated genes implicated by genome-wide association studies (GWASs). We applied scDRS to 74 diseases/traits and 1.3 million single-cell gene-expression profiles across 31 tissues/organs. Cell-type-level results broadly recapitulated known cell-type–disease associations. Individual-cell-level results identified subpopulations of disease-associated cells not captured by existing cell-type labels, including T cell subpopulations associated with inflammatory bowel disease, partially characterized by their effector-like states; neuron subpopulations associated with schizophrenia, partially characterized by their spatial locations; and hepatocyte subpopulations associated with triglyceride levels, partially characterized by their higher ploidy levels. Genes whose expression was correlated with the scDRS score across cells (reflecting coexpression with GWAS disease-associated genes) were strongly enriched for gold-standard drug target and Mendelian disease genes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
Data availability
We release our data at https://figshare.com/projects/Single-cell_Disease_Relevance_Score_scDRS_/118902 (ref. 72) (instructions at https://github.com/martinjzhang/scDRS), including GWAS summary statistics of the 74 diseases/traits, TMS FACS scRNA-seq data, reprocessed TMS FACS data (for T cells and hepatocytes), MAGMA and gold-standard gene sets and scDRS results for TMS FACS (disease scores and control scores for the 74 diseases/traits). The 16 scRNA-seq data sets were obtained as follows (15 out of 16 publicly available). The TMS FACS data and TMS droplet data17 were downloaded from the official release https://figshare.com/articles/dataset/Processed_files_to_use_with_scanpy_/8273102. The TS FACS data 20 were downloaded from the official release https://figshare.com/articles/dataset/Tabula_Sapiens_release_1_0/14267219. The Cano-Gamez & Soskic et al. data36 were downloaded from https://www.opentargets.org/projects/effectorness. The Nathan et al. data37 were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158769. The Zeisel & Muñoz-Manchado et al. data46 were downloaded from http://linnarssonlab.org/cortex/. The Zeisel et al. data51 were downloaded from http://mousebrain.org/adolescent/downloads.html. The Habib & Li et al. data50 and Habib, Avraham-Davidi, & Basu et al. data53 were downloaded from https://singlecell.broadinstitute.org/single_cell. The Ayhan et al. data55 were downloaded from https://cells.ucsc.edu/human-hippo-axis/. The Yao et al. data52 were downloaded from https://assets.nemoarchive.org/dat-jb2f34y. The Zhong et al. data54 were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119212. The Aizarani et al. data61 were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124395. The Halpern & Shenhav et al. data59 were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84498. The Richter & Deligiannis et al. data60 (annotated count matrix) were obtained via communication with the authors (raw data publicly available via links in the paper). The Taychameekiatchai et al. data are not publicly available and were provided by A.T., P.R. and B.W. MSigDB71 (v7.1) was downloaded from the official website http://www.gsea-msigdb.org/gsea/index.jsp. The Open Targets41 data were downloaded from the official website https://www.opentargets.org/.
Code availability
Software implementing scDRS and its downstream applications and a web interface for interactively exploring results of scDRS are available at https://github.com/martinjzhang/scDRS73. Code for generating all results of the paper is available at https://github.com/martinjzhang/scDRS_paper (ref. 74).
References
Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Hekselman, I. & Yeger-Lotem, E. Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nat. Rev. Genet. 21, 137–150 (2020).
Regev, A. et al. The Human Cell Atlas. elife 6, e27041 (2017).
Calderon, D. et al. Inferring relevant cell types for complex traits by using single-cell gene expression. Am. J. Hum. Genet. 101, 686–699 (2017).
Watanabe, K., Mirkov, M. U., de Leeuw, C. A., van den Heuvel, M. P. & Posthuma., D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 10, 1–13 (2019).
Bryois, J. et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of parkinson’s disease. Nat. Genet. 52, 482–493 (2020).
Hu, X. et al. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am. J. Hum. Genet. 89, 496–506 (2011).
Gormley, P. et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 48, 856–866 (2016).
Ongen, H. et al. Estimating the causal tissues for complex traits and diseases. Nat. Genet. 49, 1676–1683 (2017).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Fan, J. et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016).
Aibar, S. et al. Scenic: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
DeTomaso, D. et al. Functional interpretation of single cell similarity maps. Nat. Commun. 10, 1–11 (2019).
Cembrowski, M. S. & Spruston, N. Heterogeneity within classical cell types is the rule: lessons from hippocampal pyramidal neurons. Nat. Rev. Neurosci. 20, 193–204 (2019).
Frost, H. R. Variance-adjusted mahalanobis (VAM): a fast and accurate method for cell-specific gene set scoring. Nucleic Acids Res. 48, e94–e94 (2020).
The Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Tabula Sapiens Consortium et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171 (2014).
Skene, N. G. et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 50, 825–833 (2018).
Coleman, J. R. I. et al. The genetics of the mood disorder spectrum: genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biol. Psychiatry 88, 169–184 (2020).
Alves-Bezerra, M. & Cohen, D. E. Triglyceride metabolism in the liver. Compr. Physiol. 8, 1–8 (2017).
Guo, M. et al. Epigenetic profiling of growth plate chondrocytes sheds insight into regulatory genetic variation influencing height. elife 6, e29329 (2017).
Kemp, J. P. et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat. Genet. 49, 1468–1475 (2017).
Warren, H. R. et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat. Genet. 49, 403–415 (2017).
Chiou, J. et al. Single-cell chromatin accessibility identifies pancreatic islet cell type–and state-specific regulatory programs of diabetes risk. Nat. Genet. 53, 455–466 (2021).
De Bondt, M., Hellings, N., Opdenakker, G. & Struyf, S. Neutrophils: underestimated players in the pathogenesis of multiple sclerosis (ms). Int. J. Mol. Sci. 21, 4558 (2020).
Agarwal, D. et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 11, 1–11 (2020).
Ettle, B., Schlachetzki, J. C. M. & Winkler, J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol. Neurobiol. 53, 3046–3062 (2016).
Spitzer, S. O. et al. Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron 101, 459–471 (2019).
Huang, P. et al. Putative regulators for the continuum of erythroid differentiation revealed by single-cell transcriptome of human BM and UCB cells. Proc. Natl Acad. Sci. U S A 117, 12868–12876 (2020).
Li, A. et al. IL-33 signaling alters regulatory T cell diversity in support of tumor development. Cell Rep. 29, 2998–3008 (2019).
Abraham, C. & Cho, J. H. Inflammatory bowel disease. N. Engl. J. Med. 361, 2066–2078 (2009).
Cano-Gamez, E. et al. Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4+ T cells to cytokines. Nat. Commun. 11, 1–15 (2020).
Nathan, A. et al. Multimodally profiling memory T cells from a tuberculosis cohort identifies cell state associations with demographics, environment and disease. Nat. Immunol. 22, 781–793 (2021).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Leung, S. et al. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory t cells in autoimmune disease. Cell. Mol. Immunol. 7, 182–189 (2010).
Gutierrez-Arcelus, M. et al. Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions. Nat. Commun. 10, 1–15 (2019).
Koscielny, G. et al. Open Targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 45, D985–D994 (2017).
Freund, M. K. et al. Phenotype-specific enrichment of mendelian disorder genes near gwas regions across 62 complex traits. Am. J. Hum. Genet. 103, 535–552 (2018).
O’Connor, L. J. et al. Extreme polygenicity of complex traits is explained by negative selection. Am. J. Hum. Genet. 105, 456–476 (2019).
Zhang, H. et al. A mutation that blocks integrin α 4 β 7 activation prevents adaptive immune-mediated colitis without increasing susceptibility to innate colitis. BMC Biol. 18, 1–15 (2020).
Choy, E. H. S. et al. The effect of JAK1/JAKk2 inhibition in rheumatoid arthritis: efficacy and safety of baricitinib. Clin. Exp. Rheumatol. 37, 694–704 (2019).
Zeisel, A. et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015).
Skene, N. G. & Grant, S. G. N. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front. Neurosci. 10, 16 (2016).
Cembrowski, M. S. et al. Spatial gene-expression gradients underlie prominent heterogeneity of ca1 pyramidal neurons. Neuron 89, 351–368 (2016).
Henriksen, E. J. et al. Spatial representation along the proximodistal axis of CA1. Neuron 68, 127–137 (2010).
Habib, N. et al. Div-seq: Single-nucleus rna-seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928 (2016).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014 (2018).
Yao, Z. et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241 (2021).
Habib, N. et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017).
Zhong, S. et al. Decoding the development of the human hippocampus. Nature 577, 531–536 (2020).
Ayhan, F. et al. Resolving cellular and molecular diversity along the hippocampal anterior-to-posterior axis in humans. Neuron 109, 2091–2105.e6 (2021).
Ben-Moshe, S. & Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 16, 395–410 (2019).
Donne, R., Saroul-Aïnama, M., Cordier, P., Celton-Morizur, S. & Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 17, 391–405 (2020).
Miettinen, T. P. et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 24, 598–608 (2014).
Halpern, K. B. et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017).
Richter, M. L. et al. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nat. Commun. 12, 1–16 (2021).
Aizarani, N. et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572, 199–204 (2019).
Gazal, S. et al. Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nat. Genet. 50, 1600–1607 (2018).
Geary, R. C. The contiguity ratio and statistical mapping. incorporated statistician 5, 115–146 (1954).
Wolf, F. A., Angerer, P. & Theis, F. J. Scanpy: large-scale single-cell gene expression data analysis. Genome Biol. 19, 1–5 (2018).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc.: Ser. B (Methodol.) 57, 289–300 (1995).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 16, 1289–1296 (2019).
Traag, V. A., Waltman, L. & Eck, N. J. V. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 1–12 (2019).
Andreatta, M. et al. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 12, 1–19 (2021).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Zhang, M. J. & Hou, K. scdrs data release 030122. Figshare, https://doi.org/10.6084/m9.figshare.19312583.v1 (2022).
Zhang, M. J. & Hou, K. scdrs software v1.0.1. Zenodo, https://doi.org/10.5281/zenodo.6615722 (2022).
Zhang, M. J. & Hou, K. scdrs data analysis code v1.0.1. Zenodo, https://doi.org/10.5281/zenodo.6615791 (2022).
Sakornsakolpat, P. et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet. 51, 494–505 (2019).
Kato, K. et al. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 9, 1–14 (2018).
Acknowledgements
We thank H. Shi, K. Siewert-Rocks, T. Amariuta, X. Xu, B. J. Strober and A. Gusev for helpful suggestions. This research was conducted using the UK Biobank Resource (application number 16549). This research was funded by National Institutes of Health (NIH) grants U01 HG009379 (A.L.P. and S.R.), R01 MH101244 (A.L.P.), R37 MH107649 (A.L.P.), R01 MH115676 (A.L.P. and B.P.), U01 HG012009 (A.L.P. and S.R.) and R01 HG006399 (A.L.P.). The authors were also supported by NIH grants P01-AI148102 (S.R.), UC2-AR081023 (S.R.), R01-AR063759 (S.R.), UH2-AR067677 (S.R.), R01MH121521 (M.G.) and R01MH123922 (M.G.), and NIH National Human Genome Research Institute grants K99HG012203 (K.K.D.) and F32 HG011434-03 (K.A.J.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
M.J.Z., K.H., B.P. and A.L.P. designed the study and developed statistical methodologies. M.J.Z. and K.H. analyzed the data with assistance from K.K.D., A.O.P. and K.A.J. S.S., K.W., A.T., P.R., A.O.P., J.Z., B.W., M.G. and S.R. provided expert guidance and feedback on analysis, results and biological interpretations. M.J.Z., K.H., B.P. and A.L.P. wrote the manuscript with feedback from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Additional null simulations.
We performed null simulations for various numbers of putative disease genes (100, 500, 1,000, and 2,000 for the four columns respectively) and various types of genes to randomly sample from: all genes (first row), and top 25% genes with high expression (second row), top 25% genes with high expression variance (third row), top 25% overdispersed genes (fourth row). We considered two additional versions of scDRS: scDRS-bin-gs (binary gene sets instead of MAGMA z-score gene weights) and scDRS-adj-ctp (adjusting for cell-type proportion). For scDRS-adj-ctp, we simulated random biased gene sets (high-mean/high-variance/overdispersed) based on the balanced data (inversely weighting cells by cell-type proportion) to better match the model assumption, namely testing for excess expression relative to cells in the balanced data. In each panel, the x-axis denotes theoretical −log10 p-value quantiles and the y-axis denotes actual −log10 p-value quantiles for different methods. The 3 versions of scDRS produced well-calibrated p-values in most settings and suffered slightly inflated type I error in panels o,p, possibly because it is hard to match a large number of overdispersed putative disease genes using the remaining set of genes. In comparison, all other methods are less well-calibrated and are particularly problematic when the numbers of putative disease genes are small. Error bars denote 95% confidence intervals around the mean of 100 simulation replicates.
Extended Data Fig. 2 Additional causal simulations.
We performed three sets of causal simulations: (1) varying effect size from 5% to 50% while fixing 25% overlap (first column), (2) varying level of overlap from 5% to 50% while fixing 25% effect size (second column), (3) assigning the 528 B cells in the subsampled data to be causal (instead of the 500 randomly selected cells; varying effect size while fixing 25% overlap; third column). We report the power (first row), FDR (second row), and AUC for classifying causal from non-causal cells based on the p-values (third row). scDRS outperformed other methods under all metrics. Error bars denote 95% confidence intervals around the mean of 100 simulation replicates.
Extended Data Fig. 3 Complete results for cell type-level disease associations for 74 diseases/traits and TMS FACS 120 cell types.
Each row represents a disease/trait and each column represents a cell type (number of cells in parentheses). Heatmap colors denote the proportion of significantly associated cells (FDR < 0.1 across all cells for a given disease). Squares denote significant cell-type–disease associations (FDR < 0.05 across all pairs of the 120 cell types and 74 diseases/traits; 597 significant pairs; MC test; Methods). Cross symbols denote significant heterogeneity in association with disease across individual cells within a given cell type (FDR < 0.05 across all pairs; 273 significant pairs; MC test; Methods). Heatmap colors and cross symbols are omitted for cell-type–disease pairs with nonsignificant cell-type–disease associations. Within the blood/immune block (40 cell types and 21 diseases/traits), 136 of 264 cell-type–disease pairs with significant association also had significant heterogeneity. Within the brain block (11 cell types and 21 diseases/traits), 64 of 133 cell-type–disease pairs with significant association also had significant heterogeneity. Within the other block (69 cell types and 32 diseases/traits), 54 of 146 cell-type–disease pairs with significant association also had significant heterogeneity. We discuss the results for FEV1/FVC. We identified 20 cell types associated with FEV1/FVC (FDR < 0.05), including 5 lung cell types and 15 cell types from other tissues. They can be categorized into 5 sets of associations: (1) type II pneumocyte (2) skin-related cells (3) smooth muscle cells (4) fibroblast-and-MSC-like cells (5) pericyte-like cells. The first 4 sets of associations are consistent with a previous work75. The 5th set of pericyte associations is also plausible because pericytes are known to regulate lung morphogenesis76. We note that the cell type associations from the lung are more likely to be causal and those from the other tissues are more likely tagging the causal cell types due to shared expression. Numerical results are reported in Supplementary Table 12.
Extended Data Fig. 4 Comparison of cell type-level disease association results between TMS FACS and TMS droplet (different technologies), TS FACS (different species).
(a-c) Results for disease association at the cell type-level for TMS FACS, TMS droplet, and TS FACS for diseases and cell types in the blood/immune block (upper left) and the other cell types/diseases block (lower right) in Fig. 3 (TMS droplet and TS FACS do not contain brain data; Supplementary Tables 6,7). The plotting style is the same as Fig. 3. Heatmap colors for each cell-type–disease pair denote the proportion of significantly associated cells (FDR < 0.1); squares denote significant cell-type–disease associations (FDR < 0.05); and cross symbols denote significant heterogeneity in association with disease across individual cells within a given cell type (FDR < 0.05). Heatmap colors (>10% of cells associated) and cross symbols are omitted for cell-type–disease pairs with nonsignificant cell-type–disease associations via MC test. We matched each TMS FACS cell type using the closest cell type in the TMS droplet and TS FACS data; unmatched cell types were colored in grey. (d) Overlap of significant cell-type–disease associations between TMS FACS and TMS droplet (P = 2.8 × 10−24, two-sided Fisher’s exact test). (e) Pearson’s correlation of −log10 p-values for cell-type–disease associations between TMS FACS and TMS droplet. (f) Overlap of significant cell-type–disease associations between TMS FACS and TS FACS (P = 1.3 × 10−7, two-sided Fisher’s exact test). (g) Pearson’s correlation of −log10 p-values for cell-type–disease associations between TMS FACS and TS FACS. We determined that the results are highly consistent between TMS FACS and TMS droplet, and are reasonably consistent between TMS FACS and TS FACS. Our method is underpowered in the TS FACS data, possibly due to the smaller sample size (27K cells in TS FACS versus 110K cells in TMS FACS). The current TS FACS data corresponds to the initial data release and there will likely be more cells in future releases20.
Extended Data Fig. 5 Optimizing parameters of scDRS based on expected and unexpected control cell types across 20 traits.
We considered different versions of scDRS by varying methods for selecting (1) putative disease genes (2) weights for the disease genes (3) MAGMA window size. We considered 6 methods for selecting putative disease genes, 4 methods for selecting gene weights, and 3 MAGMA gene window sizes (Supplementary Note). We applied each version of scDRS to the subsampled TMS FACS data (20 repetitions with 10K cells each) and a curated set of 20 traits with expected and unexpected disease-critical cell types (Supplementary Table 15). For a given scDRS version and a given trait, we computed the t-statistic between cells from the expected and unexpected cell types, and divided it by the average t-statistics of results of the given trait from all data sets and all scDRS versions to correct for trait-specific baseline. We evaluated each version by first computing the mean and SE of the normalized t-statistics for a given trait across the 20 repetitions and then combining the estimates across the 20 traits via random-effect meta-analysis. We compared the performance of a pair of scDRS versions by applying the same procedure to the difference of the normalized t-statistics between the two versions. (a) Varying gene selection methods while fixing other parameters as the default. (b) Varying gene weighting methods while fixing other parameters as the default. (c) Varying MAGMA gene window size while fixing other parameters as the default. The default version was denoted in dark blue. Error bars denote 95% confidence intervals around the mean based on meta-analysis across 20 subsampled data sets and 20 traits, using procedures as described above. * denotes P < 0.05 and ** denotes P < 0.001 for significant differences relative to the default configuration; one-sided tests based on the estimated mean and CIs. Numerical results are reported in Supplementary Table 16.
Extended Data Fig. 6 Numbers of overlapping genes (upper triangle) and correlations of the scDRS disease scores across all TMS FACS cells (lower triangle) between the 26 autoimmune, brain, and metabolic traits analyzed in the main paper.
Traits are ordered via hierarchical clustering of the scDRS score correlation and the clustering dendrogram was provided. The level of gene set overlap is moderate. scDRS disease score correlations distinguish diseases/traits from the 3 categories as well as subgroups of diseases/traits in the same category.
Extended Data Fig. 7 Additional results on disease gene prioritization.
(a–j) Comparison to alternative disease gene prioritization methods for the 10 autoimmune diseases. The first row shows levels of excess overlap between the prioritized disease genes and the gold standard gene sets while the second row shows the corresponding −log10 p-values for excess overlap. Each dot corresponds to a disease, the y-axis shows results for the proposed prioritization method (correlating gene expression levels with the scDRS disease score across all TMS FACS cells), and the x-axis shows results from comparison methods, including (from left to right) top 1,000 MAGMA genes, top 1,000 genes specifically expressed in T cells (vs. the rest of cells in TMS FACS), prioritization based on correlation across T cells (instead of all TMS FACS cells), prioritization based on correlation across CD4+ T cells (instead of all TMS FACS cells), and prioritization based on correlation across CD8+ T cells (instead of all TMS FACS cells). (k-l) Overlap with drug target genes for 27 diseases. (m-n) Overlap with Mendelian disease genes for 45 diseases. The median ratio of −log10 p-values and (excess overlap − 1) between the y- and x-values (median of ratios) was provided in the figure title. P-values are based on two-sided Fisher’s exact tests.
Extended Data Fig. 8 Complete results of correlations between scDRS disease scores and inferred spatial coordinates across CA1 pyramidal neurons in 7 single-cell data sets (extending results in Fig. 5b).
(a) Results for regressing the scDRS disease scores against the inferred spatial coordinates for each disease/trait and each inferred spatial coordinate. Color represents the t-statistics and stars represent significant associations (* denotes P < 0.05 and ** denotes P < 0.005, one-sided MC test; Methods). For clarification, Zeisel & Muñoz-Manchado et al. refers to the data from Zeisel & Muñoz-Manchado et al. 2015 Science46 and Zeisel et al. refers to the data from Zeisel et al. 2018 Cell51. (b) Summary of results in panel a. Heatmap color represent the average t-statistics across the 7 brain-related diseases/traits (excluding height) for each data set and stars represent significant associations by combining p-values across data sets using Fisher’s combined probability test. (c) Summary of the association between brain-related diseases and the inferred spatial coordinates for the mouse and human data sets in panel b.
Extended Data Fig. 9 Complete results of joint regression analysis for GWAS metabolic traits and putative zonated metablic processes across the 6 data sets (extending results in Fig. 5d).
(a, b) Results for the 9 metabolic traits and height, a negative control trait. The polyploidy score (panel a) and both the pericentral and periportal score (panel b) were consistently associated with the 9 metabolic traits across the data sets. The strong association (P < 0.005) between the pericentral score and height in the Aizarani et al. data may be because that we inferred the pericentral score using mouse gene signatures, which are less conserved in human (as also mentioned in the original paper61). (c, d) Results for the 8 metabolic pathways. Overall, as shown in panel d, the pericentral score was associated with pericentral-specific pathways (first 4 rows) while the periportal score was associated with periportal-specific pathways (last 4 rows). * denotes P < 0.05 and ** denotes P<0.005 based on one-sided MC tests.
Supplementary information
Supplementary Information
Supplementary Note, Supplementary Tables 1–27 and Supplementary Figures 1–27.
Supplementary Table 1
Supplementary data tables.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, M.J., Hou, K., Dey, K.K. et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nat Genet 54, 1572–1580 (2022). https://doi.org/10.1038/s41588-022-01167-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-022-01167-z
This article is cited by
-
Decoding the spatiotemporal regulation of transcription factors during human spinal cord development
Cell Research (2024)
-
Single-cell analysis reveals the spatial-temporal expression of genes associated with esophageal malformations
Scientific Reports (2024)
-
Leveraging single-cell ATAC-seq and RNA-seq to identify disease-critical fetal and adult brain cell types
Nature Communications (2024)
-
Genetic variation across and within individuals
Nature Reviews Genetics (2024)
-
Drug targeting in psychiatric disorders — how to overcome the loss in translation?
Nature Reviews Drug Discovery (2024)
