
Abstract
Characterization of the genetic landscape of Alzheimer’s disease (AD) and related dementias (ADD) provides a unique opportunity for a better understanding of the associated pathophysiological processes. We performed a two-stage genome-wide association study totaling 111,326 clinically diagnosed/‘proxy’ AD cases and 677,663 controls. We found 75 risk loci, of which 42 were new at the time of analysis. Pathway enrichment analyses confirmed the involvement of amyloid/tau pathways and highlighted microglia implication. Gene prioritization in the new loci identified 31 genes that were suggestive of new genetically associated processes, including the tumor necrosis factor alpha pathway through the linear ubiquitin chain assembly complex. We also built a new genetic risk score associated with the risk of future AD/dementia or progression from mild cognitive impairment to AD/dementia. The improvement in prediction led to a 1.6- to 1.9-fold increase in AD risk from the lowest to the highest decile, in addition to effects of age and the APOE ε4 allele.
Similar content being viewed by others


Main
AD is the most common form of dementia. The heritability is high, estimated to be between 60% and 80%1. This strong genetic component provides an opportunity to determine the pathophysiological processes in AD and to identify new biological features, new prognostic/diagnostic markers and new therapeutic targets through translational genomics. Characterizing the genetic risk factors in AD is therefore a major objective; with the advent of high-throughput genomic techniques, a large number of putative AD-associated loci/genes have been reported2. However, much of the underlying heritability remains unexplained. Hence, increasing the sample size of genome-wide association studies (GWASs) is an obvious solution that has already been used to characterize new genetic risk factors in other common, complex diseases (e.g., diabetes).
GWAS meta-analysis
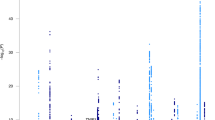
The European Alzheimer & Dementia Biobank (EADB) consortium brings together the various European GWAS consortia already working on AD. A new dataset of 20,464 clinically diagnosed AD cases and 22,244 controls has been collated from 15 European countries. The EADB GWAS results were meta-analyzed with a proxy-AD GWASs of the UK Biobank (UKBB) dataset. The UKBB’s proxy-AD designation is based on questionnaire data in which individuals are asked whether their parents had dementia. This method has been used successfully in the past3 but is less specific than a clinical or pathological diagnosis of AD; hence, we will refer to these cases as proxy AD and related dementia (proxy-ADD). EADB stage I (GWAS meta-analysis) was based on 39,106 clinically diagnosed AD cases, 46,828 proxy-ADD cases (as defined in the Supplementary Note), 401,577 controls (Supplementary Tables 1 and 2) and 21,101,114 variants that passed our quality control (Fig. 1; see Supplementary Fig. 1 for the quantile–quantile plot and genomic inflation factors). We selected all variants with a P value below 1 × 10−5 in stage I. We defined nonoverlapping regions around these variants, excluded the region corresponding to APOE and examined the remaining variants in a large follow-up sample that included AD cases and controls from the ADGC, FinnGen and CHARGE consortia (stage II; 25,392 AD cases and 276,086 controls). A signal was considered as significant on the genome-wide level if it (1) was nominally associated (P ≤ 0.05) in stage II, (2) had the same direction of association in the stage I and II analyses and (3) was associated with the ADD risk with P ≤ 5 × 10−8 in the stage I and stage II meta-analysis. Furthermore, we applied a PLINK clumping procedure4 to define potential independent hits within the stage I results (Methods). After validation by conditional analyses (Supplementary Note and Supplementary Tables 3 and 4), this approach enabled us to define 39 signals in 33 loci already known to be associated with the risk of developing ADD3,5,6,7,8,9,10 and identify 42 loci defined as new at the time of analysis (Tables 1 and 2, Supplementary Table 5 and Supplementary Figs. 2–29). Of the 42 new loci, 17 had P ≤ 5 × 10−8 in stage I and 25 were associated with P ≤ 5 × 10−8 after follow-up (stage I and stage II meta-analysis, including the ADGC, CHARGE and FinnGen data). We also identified 6 loci with P ≤ 5 × 10−8 in the stage I and stage II analysis but with P > 0.05 in stage II (Supplementary Table 6). It is noteworthy that the magnitude of the associations in stage I did not change substantially if we restricted the analysis to clinically diagnosed AD cases (Supplementary Table 7 and Supplementary Fig. 30). Similarly, none of the signals observed appeared to be especially driven by the UKBB data (Supplementary Table 7 and Supplementary Figs. 2–29). Nine of these loci (APP, CCDC6, GRN, LILRB2, NCK2, TNIP1, TMEM106B, TSPAN14 and SHARPIN) were recently reported in three articles using part of the GWAS data included in our study11,12,13. We also generated a detailed analysis of the human leukocyte antigen (HLA) locus on the basis of the clinically diagnosed AD cases (Supplementary Tables 8 and 9, Supplementary Figs. 31 and 32 and Supplementary Note).

P values are two-sided raw P values derived from a fixed-effect meta-analysis. Variants with a P value below 1 × 10−36 are not shown. Loci with a genome-wide significant signal are annotated (known loci in black and new loci in red). Variants in new loci are highlighted in red. The red dotted line represents the genome-wide significance level (P = 5 × 10−8), and the black dotted line represents the suggestive significance level (P = 1 × 10−5).
Genetic overlap with other neurodegenerative diseases
We tested the association of the lead variants within our new loci with the risk of developing other neurodegenerative diseases or AD-related disorders (Supplementary Fig. 33 and Supplementary Tables 10–12). We also performed more precise colocalization analyses (using Coloc R package, https://cran.r-project.org/web/packages/coloc/index.html) for five loci known to be associated with Parkinson’s disease (IDUA and CTSB), types of frontotemporal dementia (TMEM106B and GRN) and amyotrophic lateral sclerosis (TNIP1) (Supplementary Tables 13 and 14). The IDUA signal for Parkinson’s disease was independent of the signal in ADD (coloc posterior probability (PP)3 = 99.9%), but we were not able to determine whether the CTSB signals colocalized. The TMEM106B and GRN signals in frontotemporal lobar degeneration with TAR DNA-binding protein (TDP-43) inclusions (frontotemporal lobar degeneration TDP) probably share causal variants with ADD (coloc PP4 = 99.8% and coloc PP4 = 80.1%, respectively). Lastly, we were not able to determine whether the TNIP1 signals colocalized for ADD and amyotrophic lateral sclerosis.
Pathway analyses
Next, we sought to perform a pathway enrichment analysis on the stage I association results to gain better biological understanding of this newly expanded genetic landscape for ADD. Ninety-three gene sets were still statistically significant after correction for multiple testing (q ≤ 0.05; Methods and Supplementary Table 15). As described previously, the most significant gene sets are related to amyloid and tau5; other significant gene sets are related to lipids, endocytosis and immunity (including macrophage and microglial cell activation). When restricting this analysis to the meta-analysis based on the clinically diagnosed AD cases, 54 gene sets were significant (q ≤ 0.05). Of these 54 gene sets, 33 reached q ≤ 0.05 in the stage I analysis and all reached P ≤ 0.05. This indicates that the inclusion of proxy-ADD cases does not cause disease-relevant biological information to be missed and underlines the additional power of this type of analysis.
We next performed a single-cell expression enrichment analysis by using the average gene expression per nucleus (Av. Exp.) data in the human Allen Brain Atlas (49,495 nuclei from 8 human brains). Only the microglial expression reached a high level of significance (P = 1.7 × 10−8; Supplementary Table 16); greater expression corresponded to a more significant association with ADD. After adjusting for microglial Av. Exp., the remaining associations became nonsignificant; this indicates that microglial Av. Exp. drives all the other cell-type associations. These results were observed whatever the brain region studied (Supplementary Table 16). A similar result was observed using a mouse single-cell dataset14 (Supplementary Table 17 and Supplementary Note).
Lastly, we looked at whether the relationship between an elevated microglia Av. Exp. and a genetic association with the ADD risk was specific to particular biological processes (Supplementary Table 18) by analyzing the interaction between microglia Av. Exp. and pathway membership in MAGMA15. Of the five most significant interaction signals (q ≤ 10−3), two were directly associated with endocytosis processes (GO:0006898 and GO:0031623); this suggested a functional relationship between microglia and endocytosis, which is known to be involved in phagocytosis (Supplementary Table 18). It is noteworthy that we also detected an interaction between GO:1902991 (regulation of amyloid precursor protein (APP) catabolic process) and the gene expression level in microglia (q = 1.4 × 10−3; Supplementary Table 18). Even though these data suggest a functional relationship between microglia and APP/amyloid beta (Aβ) peptide pathways, this observation reinforces the likely involvement of microglial endocytosis in AD, a mechanism that is also strongly involved in APP metabolism16. Of note, there are overall similarities in the interaction effects of human and mouse microglia expression with genes in biological pathways of relevance to the AD genetic risk (Supplementary Table 18 and Supplementary Note).
Gene prioritization
We next attempted to identify the genes most likely to be responsible for the association signal with ADD at each new locus. To this end, we studied the downstream effects of ADD-associated variants on molecular phenotypes (i.e., expression, splicing, protein expression, methylation and histone acetylation) in various cis-quantitative trait locus (cis-QTL) catalogues from AD-relevant tissues, cell types and brain regions. We investigated the genetic colocalization between association signals for the ADD risk and those for the molecular phenotypes and the association between the ADD risk and these phenotypes by integrating cis-QTL information into our ADD GWAS. Moreover, we considered the lead variant annotation (the allele frequency, protein-altering effects and nearest protein-coding gene) and a genome-wide, high-content short interfering RNA screen for APP metabolism17. Based on this evidence, we developed a systematic gene prioritization strategy that yielded a total weighted score of between 0 and 100 for each gene (Supplementary Fig. 34 and Supplementary Note). This score was used to compare and prioritize genes in the new loci within 1 Mb upstream and 1 Mb downstream of the lead variants. Genes either were ranked as tier 1 (greater likelihood of being the causal risk gene responsible for the ADD signal) or tier 2 (lower likelihood and the absence of a minimum level of evidence as a causal risk gene) or were not ranked.
From all newly identified loci, this gene prioritization yielded 31 tier 1 genes and 24 tier 2. The 55 prioritized genes, the details of the analyses and the supporting evidence are summarized in Fig. 2a and the Supplementary Note (Supplementary Tables 19–30 and Supplementary Figs. 35–45). Among the 31 tier 1 genes, we observed that 25 of these genes were the only prioritized gene in their respective locus. For the remaining 6 tier 1 genes, we also found tier 2 genes in their respective locus. We also identified five loci containing several tier 2 prioritized genes. In one of these loci, locus 39 (L39), the tier 2 prioritized gene LILRB2 had strong additional support from published literature (Supplementary Note). In five loci, our prioritization score did not identify sufficient molecular evidence to prioritize genes with exception of being the nearest gene (L10, L12, L13, L14 and L32). Finally, we excluded the complex IGH cluster (L27) from gene prioritization analyses due to genomic complexity of the telomeric locus as a consequence of known fusion events18.

a, Summary of weighted scores for each evidence category for the prioritized genes in the 42 new genome-wide-significant loci. Using our gene prioritization method, we considered the genes within 1 Mb of each new lead variant and prioritized a total of 55 genes in 42 new loci at two different confidence levels (31 tier 1 genes and 24 tier 2 genes). The leftmost squares indicate the new locus index number. The different types of evidence are colored according to the seven different domains to which they belonged. Weighted scores for each evidence category are rescaled to a 0–100 scale, and the proportions of mean human brain cell-type-specific expression for each gene are also rescaled to a 0–100 scale; darker colors represent higher scores or higher expression proportions. Tier 1 genes are shown in dark green, and tier 2 genes are shown in light green. Only tier 1 and tier 2 genes are shown for each locus. Supplementary Fig. 35 shows full results. MAFs and CADD (v1.6) PHRED scores for rare and/or protein-altering rare variants are labeled in white within the respective squares. b, Pathway enrichment analysis based on the tier 1 gene list. Only the ten strongest associations (according to STRING software) are presented here. coloc, colocalization; eQTL, expression QTL; eTWAS, expression transcriptome-wide association study; GO, Gene Ontology; haQTL, histone acetylation QTL; Mon. Mac., monocytes and macrophages; sTWAS, splicing transcriptome-wide association study; m/haQTL, methylation/histone acetylation QTL; sQTL, splicing QTL; FDR, false discovery rate.
We highlight two examples, L18 and L23. In L18, the lead variant, rs76928645 (MAF = 10%), is intergenic and is located more than 100 kb downstream or upstream of the two nearest protein-coding genes (SEC61G and EGFR, respectively). Our gene prioritization analyses suggested that EGFR was the only risk gene (Fig. 3). We found that both the lead variant (rs76928645) and the other nearby variants in linkage disequilibrium (LD) are significant expression QTLs (eQTLs) for regulating EGFR expression downstream. The eQTL signals in brain strongly colocalized with the GWAS signal (with eQTL coloc PP4s of 98.3% in the temporal cortex (TCX) and 99.5% in the dorsolateral prefrontal cortex (DLPFC)). Accordingly, the fine-mapped expression transcriptome-wide association study (eTWAS) associations (Fine-mapping Of CaUsal gene Sets (FOCUS) posterior inclusion probability (PIP) = 1; eTWAS P = 6.9 × 10−9, eTWAS Z = + 5.8 in the TCX; eTWAS P = 3.1 × 10−11, eTWAS Z = + 6.6 in the DLPFC) indicated that genetic downregulation of EGFR expression is associated with a lower ADD risk (Fig. 3; Supplementary Tables 22, 24 and 26; and Supplementary Figs. 36a, 39 and 41).

a, The regional plot of the new SEC61G locus (L18) shows the EADB GWAS stage I (n = 487,511) ADD association signal within 200 kb of the intergenic lead variant, rs76928645 (the two closest protein-coding genes, SEC61G and EGFR, are more than 100 kb from the lead variant), together with the eQTLs in the same region identified for SEC61G and EGFR expression separately in the TCX (MayoRNAseq TCX eQTL catalog based on n = 259 individuals). The rs7692864 lead variant is shown in purple, and LD r2 values (calculated for the EADB Trans-Omics for Precision Medicine (TOPMed) dataset (n = 42,140) with respect to the lead variant) are indicated on a color scale. y axis, −log10 for the GWAS or eQTL P value; x axis, hg38 genomic position on chromosome 7. b, Colocalization between the EGFR eQTL signal (MayoRNAseq TCX, n = 259 individuals) and the EADB GWAS stage I (n = 487,511) signal (eQTL coloc PP4 = 98.3%); with the significant eTWAS association (eTWAS P = 6.9 × 10−9 and eTWAS Z = 5.8) and fine-mapped (FOCUS PIP = 1) eTWAS association in the same catalog. y axis, eQTL −log10(P) value; x axis, GWAS −log10(P) value. LD r2 values and color scales are as in a. c, The eQTL violin plot shows a significant association (eQTL P = 3 × 10−18) between the rs76928645 lead variant genotype and EGFR expression in the TCX (MayoRNAseq TCX, n = 259 individuals), where the protective allele T is associated with lower EGFR expression (eQTL β, −0.39). Each data point represents a sample whose normalized EGFR expression value is indicated on the y axis, and the rs76928645 genotype information is indicated on the x axis. The lower and upper hinges of the boxes represent the first and third quantiles, the whiskers extend 1.5 times the interquartile range from the hinges and the line represents the median.
In L23, we observed numerous eQTL-GWAS and methylation QTL (mQTL)-GWAS hits for TSPAN14 that support the hypothesis that increased brain expression of TSPAN14 is associated with increased ADD risk. We also identified several splice junctions in TSPAN14 whose genetic regulation signals in lymphoblastoid cell lines (LCLs) and brain colocalized with the ADD association signal. These splice junctions were also associated with ADD risk (Fig. 4, Supplementary Tables 22–28 and Supplementary Figs. 36–42 and 44c). As three of these splice junctions were related to new complex cryptic splicing events that were predicted to result in two cryptic exons not previously described in known TSPAN14 transcripts (based on GENCODE v38), we designed a long-read single-molecule (Nanopore) sequencing experiment (Supplementary Note) to validate these cryptic exons on a total of 93 complementary DNA (cDNA) samples derived from LCLs, frontal cortex and hippocampus and consequently validated those cryptic exons (Fig. 4). All three of the validated cryptic splicing events occur within the ADAM10-interacting domain of TSPAN14. Cryptic exon 1 is at least 45 bp long, and cryptic exon 2 is 118 bp long.

a, Splicing QTL (sQTL)-GWAS integration results. Known TSPAN14 transcripts (GENCODE v38; green, coding sequences; gray, noncoding) plotted with −log10(P) for (1) EADB GWAS stage I (n = 487,511) signal (black), (2) sQTL signal for chr10:80509471–80510106 junction (supporting cryptic exon 1) in the EADB Belgian LCL sQTL catalog (n = 70 individuals, blue) and (3) sQTL signal for chr10:80512269–80512719 junction in the MayoRNAseq TCX sQTL catalog (n = 259 individuals, red); hg38 genomic position is shown above. LCL and brain-based sQTL coloc and sTWAS analyses associate ADD risk with these junctions that suggest cryptic splicing within ADAM10-interacting domain of TSPAN14 (magenta), which was predicted to result in two cryptic exons. b, Long-read sequencing validation of TSPAN14 cryptic exons. Nanopore sequencing results (Supplementary Note) in the zoomed-in region of chr10:80506973–80516400 (cumulative coverage in log10 scale). Pooled LCL cDNA sample sequenced for cDNA Amplicon2 shown in blue. cDNA Amplicon1 was sequenced on biologically independent hippocampal (HPC; n = 16, red), frontal cortex (FC; n = 18, pink) and LCL (n = 59, orange) cDNA samples. Green, canonical exons (8–12); dotted black lines, canonical splicing; blue, cryptic exon 1 (>45 bp); red, cryptic exon 2 (118 bp). All annotated junctions use canonical splice donor (GT) and acceptor (AG) sites. c,d, sQTL-GWAS colocalization plots for chr10:80509471–80510106 (supporting cryptic exon 1) in the EADB Belgian LCL sQTL catalog (n = 70 individuals) (c) and chr10:80512269–80512719 (supporting cryptic exon 2) in the MayoRNAseq TCX sQTL catalog (n = 259 individuals) (d). sQTL signals for the two junctions colocalize with ADD signal (PP4s of 98.8% and 97.4%, respectively), and sTWAS associates with increased preference for the cryptic splicing with decreased ADD risk (sTWAS P = 6.28 × 10−12 and 1.6 × 10−13, sTWAS Z = −6.9 and −7.4, respectively). y axis, sQTL −log10(P); x axis, EADB GWAS stage I −log10(P). LD r2 values calculated within EADB-TOPMed dataset (n = 42,140) based on the lead variant rs6586028 (purple) are indicated on a color scale.
Lastly, we used STRING v11 (ref. 19) to analyze protein–protein interaction for (1) previously known AD genes from GWASs, (2) our prioritized new genes (tier 1 in Fig. 2a and Supplementary Table 20) and (3) a combination of the two (Supplementary Note). The largest networks contained 14, 8 and 30 proteins, respectively (Supplementary Fig. 46). These networks were larger than would be expected by chance (respectively, P < 2 × 10−5, P = 2.8 × 10−3 and P < 2 × 10−5 based on comparison with 50,000 randomly simulated protein lists matched for the number of proteins and the total number of interactions for each protein). Notably, the number of interactions between our prioritized genes and previously known genes is also significantly greater than would be expected (P < 1 × 10−4), indicating that the newly prioritized genes are biologically relevant in AD. No such enrichment (P = 0.88) was observed for the remaining genes in the new loci, again highlighting the value of our prioritization approach.
We next performed a pathway enrichment analysis of the tier 1 genes using STRING. We found that several gene sets linked to the immune system remained statistically significant after correction for multiple testing (Fig. 2b and Supplementary Table 31), especially regulation of the tumor necrosis factor (TNF)-mediated signaling pathway (GO:0010803). We report the potential genetic implication of the linear ubiquitin chain assembly complex (LUBAC), which is a major regulator of the aforementioned signaling pathway20. Two of the LUBAC’s three complements are encoded by the new tier 1 prioritized genes SHARPIN and RBCK1, and the complex’s function is directly regulated by OTULIN (also a new tier 1 prioritized gene).
GRS
We next looked at whether the genetic ADD burden (as measured by a genetic risk score (GRS)) generated from our genome-wide significant variants (n = 83, excluding APOE; Supplementary Table 32) might influence the rate of conversion to AD in (1) individuals from several prospective, population-based cohorts and (2) patients with mild cognitive impairment (MCI) in prospective memory clinic studies (Supplementary Table 33). We used Cox regression models to assess the association after adjustment for age at baseline, sex, the number of APOE-ε4 and APOE-ε2 alleles, and genetic principal components (PCs).
In population-based cohorts with clinically diagnosed AD cases, the GRS was significantly associated with conversion to AD; this was shown in a fixed-effect meta-analysis (hazard ratio (HR) (95%CI) per average risk allele = 1.076 (1.064–1.088), P = 9.2 × 10−40; Fig. 5 and Supplementary Table 34). Likewise, the GRS was significantly associated with AD conversion in patients with MCI (HR = 1.056 (1.040–1.072), P = 2.8 × 10−12; Fig. 5 and Supplementary Table 35). Furthermore, we found that the GRS association increased significantly when the new variants discovered in the present study were added to the previously described variants (Supplementary Table 36) for both population-based studies (HR = 1.052 (1.037–1.068), P = 1.5 × 10−11) and MCI cohorts (HR = 1.034 (1.013–1.055), P = 1.4 × 10−3).

a,b, Meta-analysis results of the association between the GRS and the risk of progression to AD in population-based cohorts (n = 17,545 independent samples) (a) and MCI cohorts (n = 4,114 independent samples) (b). Data are presented as HR together with 95% CIs derived from Cox regression analyses for each individual cohort. HRs indicate the effect of the GRS as the increment in the AD risk associated with each additional average risk allele in the GRS. Null hypothesis testing is based on a meta-analysis of individual cohort effects using fixed effects (FE) and random effects (RE) models. Resulting HRs and 95% CIs and the respective Z test and associated two-sided P value are shown at the bottom of the figure. Heterogeneity between cohorts is indicated by the I2 index together with the respective Cochran’s Q statistic (distributed as χ² statistic), associated degrees of freedom and P value. 3C, Three-City Study; AgeCoDe, German study on aging cognition and dementia; AMC, additional, independent memory clinic cohort from Fundacio ACE; DCN, German Dementia Competence Network study; FACE, Fundacio ACE memory clinic cohort; FHS, Framingham Heart Study; HAN, BALTAZAR multicenter prospective memory clinic study; MAS, Sydney Memory and Ageing Study; RS1, Rotterdam Study first cohort; RS2, Rotterdam Study second cohort; VITA, Vienna Transdanube Aging study; UAN, memory clinic cohort from the Hospital Network Antwerp; UHA, University of Halle memory clinic cohort; ZIM, Heidelberg/Mannheim memory clinic sample.
Importantly, the results of our meta-analysis suggest that the risk of conversion to AD rises with the number of risk alleles from non-APOE risk variants in the GRS by 1.9-fold in population-based cohorts (HR = 1.93 (1.75–2.13); Fig. 5) and 1.6-fold in MCI cohorts (HR = 1.63 (1.42–1.87); Fig. 6) on top of effects of age and the APOE ε4 allele. These observations result from the comparison of hypothetical individuals with a GRS value at the first decile of the distribution versus those with a GRS value at the ninth decile (Fig. 6). With regard to APOE, carrying an additional APOE-ε4 allele was associated with a slightly higher increase in the AD risk in population-based cohorts (HR = 2.19 (2.03–2.37)) and MCI cohorts (HR = 1.90 (1.73–2.07)). There was no interaction between the GRS and the number of APOE-ε4 alleles (Supplementary Table 37).

a,b, Representative plots of the progression to AD over 10 years in the population-based 3C study (a) and the progression from MCI to AD over 5 years in the Fundació ACE cohort (b). The figures show the probabilities of conversion (survival probabilities) to AD (y axes) for a hypothetical participant with average covariates (mean values for age and PCs, and the mode for sex and APOE) and a GRS at the first (lowest) decile (in blue) or a GRS at the ninth (highest) decile (red). The shaded regions correspond to the 95% CI.
In an MCI cohort setting, this effect of the GRS corresponds to a median AD conversion probability within 3 years of 21.9% in patients with a GRS below the first decile (range, 4.1–34.9%) and 37.5% (range, 10.8–56.2%) in patients with a GRS above the ninth decile. There was a consistent increase in probability between these deciles in all cohorts (median (range), 13.8% (6.6–25.0%); Supplementary Table 38).
To better define the GRS discriminative ability regarding AD conversion, we assessed the improvements in three indices of predictive performance after adding the GRS to a Cox model containing age, sex, PCs and the number of APOE-ε4 and APOE-ε2 alleles as covariates (Supplementary Tables 34 and 35). We found a small but consistent increase in the discrimination between AD converters and nonconverters, as indicated by the concordance index (C-index) in population-based cohorts (Δ5years-C-indexfixed-effects = 0.002 (0.0004–0.004)) and MCI cohorts (Δ3years-C-indexfixed-effects = 0.007 (0.001–0.012)). This finding was further supported by small-to-moderate increases in the continuous NRI (net reclassification improvement) index in population-based cohorts (NRI5year-fixed-effects = 0.248 (0.159–0.336)) and MCI cohorts (NRI3year-fixed-effects = 0.232 (0.140–0.325)); this indicates that the risk assignment is more appropriate to individuals when the GRS is taken into account21. Furthermore, an increase in the index of prediction accuracy (IPA) was observed in all of the population-based cohorts (average Δ5years-IPAfixed-effects = 0.29% (0.23%–0.35%)) and all but one of the MCI cohorts (average Δ3years-IPAfixed-effects = 1.53% (1.31%–1.76%)), indicating an overall improvement in predictive performance. As expected, the amount of improvement in this index varied greatly from one cohort to another, given its dependency on incidence rates. The value of adding the new genetic variants was emphasized by the fact that effect sizes (as measured by the indices of predictive ability) were lower when only previously known AD risk variants were included in the GRS (Supplementary Table 39).
The results were similar when we (1) computed indices for other follow-up time points, (2) applied a random effects meta-analysis, (3) considered conversion to all-cause-dementia as the outcome and (4) excluded the Framingham Heart Study (FHS), as it was part of the stage II of the GWAS from which ORs for PRS computation were extracted (Supplementary Tables 34–44 and Supplementary Fig. 47).
Discussion
Our meta-analysis combined a large, new case–control study with previous GWASs. We identified 75 independent loci for ADD; 33 had been reported previously, and 42 correspond to new signals at the time of this analysis. The prioritized genes and their potential impact on the pathophysiology of AD are described in the Supplementary Note.
Our pathway enrichment analyses removed ambiguities concerning the involvement of tau-binding proteins and APP/Aβ peptide metabolism in late-onset AD processes at a much higher level than had been described previously5. It is noteworthy that new genetic risk factors are often first evaluated in the context of known pathways; many new research approaches were developed to systematically characterize putative links among APP metabolism, tau function and ADD genetic risk factors22,23. This approach can lead to circular reasoning and thus artificial enrichment in specific processes. However, we implicate ADAM17, a gene whose protein product is known to carry α-secretase activity as ADAM10 (ref. 24). This observation suggests that the nonamyloidogenic pathway for APP metabolism might be deregulated in AD. In addition to APP, we also identified six highly plausible prioritized (tier 1) genes (ICA1L, DGKQ, ICA1, DOC2A, WDR81 and LIME1) that are likely to modulate the metabolism of APP.
These pathway enrichment analyses also confirmed the involvement of innate immunity and microglial activation in ADD (Supplementary Table 15). Our single-cell expression enrichment analysis also highlighted genes expressed in microglia (Supplementary Tables 16 and 17). Indeed, three of our prioritized (tier 1) genes (RHOH, BLNK and SIGLEC11) and two of our tier 2 genes (LILRB2 and RASGE1FC) appeared to be mainly expressed in microglia (>90% relative to the total expression summed across cell types; Fig. 2a and Supplementary Table 45). Importantly, SIGLEC11 and LILRB2 have already been linked to Aβ peptides/amyloid plaques25,26.
Here, we also provide genetic evidence of the LUBAC’s potential implication in ADD. Two of the LUBAC’s three complements are encoded by SHARPIN and RBCK1, and the LUBAC is regulated by OTULIN; all three genes were found to be high-confidence, prioritized risk genes in our study. The LUBAC is the only E3 ligase known to form linear ubiquitin chains de novo through ubiquitin’s N-terminal methionine. The complex has mostly been studied in the context of inflammation, innate immunity and defense against intracellular pathogens. For instance, the LUBAC is reportedly essential for NLRP3 inflammasome activation27 and thus acts as a key innate immune regulator28. In turn, the NLRP3 inflammasome is essential for the development and progression of Aβ pathology in mice29 and may drive tau pathology through Aβ-induced microglial activation30. The LUBAC is also reportedly involved in autophagy, and linear ubiquitin chain modifications of TDP-43-positive neuronal cytoplasmic inclusions have been described as potential inducers of autophagic clearance31. Lastly, the LUBAC has been studied as a regulator of TNF-α signaling in particular20.
Interestingly, the TNF-α signaling pathway was also flagged by other genetic findings in our study (Supplementary Fig. 48). For example, ADAM17 (also known as TNF-α-converting enzyme) is of pivotal importance in the activation of TNF-α signaling32. For TNIP1, its gene product (TNF-α-induced protein 3-interacting protein 1) is involved in the inhibition of the TNF-α signaling pathway and nuclear factor κB activation/translocation33. Additional signal related to TNF-α is the one found at SPPL2A (one of the 33 confirmed loci). The protein encoded by SPPL2A is involved in noncanonical shedding of TNF-α34, and PGRN has been described as a TNF receptor ligand and an antagonist of TNF-α signaling35. Several lines of evidence had linked the inhibition of TNF-α signaling with reduction of both Aβ and tau pathologies in vivo36,37. Although a potential inflammatory connection has been suggested for TNF-α through the activation of NLRP3 inflammasome38, the TNF-α signaling pathway is also involved in many other brain physiological functions (e.g., synaptic plasticity in neurons) and pathophysiological processes (e.g., synapse loss) in the brain39. Furthermore, the involvement of the TNF-α signaling pathway and the LUBAC might be important in cell types other than microglia in AD. It is important to note that six of our prioritized (tier 1) genes (ICA1L, EGFR, RITA1, MYO15A, LIME1 and APP) are expressed at a low level in microglia (<10%, relative to the total expression summed across cell types; Supplementary Table 45), emphasizing that ADD results from complex crosstalk between different cell types in the brain23,40. It is also noteworthy that the EGFR pathway is known to interact with the TNF-α signaling pathway41, which suggests interplay between the two signaling pathways during the ADD development.
A better understanding of the etiology of ADD might also result from the observation that the risks of developing ADD and frontotemporal dementia are associated with the same causal variants in GRN and TMEM106B. This association might be due to the misclassification of clinical diagnosis of AD and the presence of proxy-ADD cases in the UKBB. However, GRN and TMEM106B have also been linked to brain health and many other neurodegenerative diseases. For instance, GRN and TMEM106B are reportedly potential genetic risk factors for differential aging in the cerebral cortex42 and cognitive impairment in amyotrophic lateral sclerosis43 and Parkinson’s disease44,45. Lastly, both GRN and TMEM106B have already been associated with neuropathological features of AD46,47,48. Taken as a whole, these data may thus emphasize a potential continuum between neurodegenerative diseases in which common pathological mechanisms are driven by GRN and TMEM106B. Interestingly, both GRN and TMEM106B are reported to be involved in defective endosome/lysosome trafficking/function49,50, a defect that is also observed in AD.
By applying a GRS derived from all the genome-wide-significant variants discovered in this study, we identified an association with the risk of incident AD in prospective population-based cohorts and with the risk of progression over time from MCI to AD (Fig. 5 and Supplementary Table 33). In patients with MCI, previous associations of AD risk with a GRS built on previously known genetic AD risk variants has been inconsistent51. It is important to note that the GRS has an impact on the AD risk in addition to that of age and that the GRS’s effect is independent of APOE status. With a view to translating genetic findings into preventive measures and personalized medicine, we also sought to provide the GRS’s added value for risk prediction by calculating the discriminative capacity through three different indices. Overall, the indices suggested that the effect size for the association between the GRS and AD was small but significant. Despite this modest effect, the inclusion of the GRS into the predictive model consistently improved the assignment of the risk of progression, as expressed by the net reclassification improvement (NRI) index21. Importantly, the cumulative improvements in risk prediction (due to inclusion of the new variants in the GRS) led to a 1.6- to 1.9-fold increase in the AD risk from the lowest to the highest decile, in addition to the effects of age and APOE status. We also showed that in addition to known risk variants, the new risk variants identified in the present study are significantly associated with progression to AD. The results of future GWASs are expected to further improve AD-risk prediction. Hence, the GRS will help to sharpen the threshold that differentiates between people at risk of progressing to dementia and those who are not.
A recent study estimated that fewer than 100 causal common variants may explain the entire AD risk52; if that estimate is correct, then our study might have already characterized a large proportion of this genetic component due to common variants. However, several reasons strongly underscore the need for additional efforts to fully characterize the still-missing AD genetic component. First, it is probable that additional, yet-unknown loci bear common variants modulating the risk for AD. Second, identification of rare variants with very low frequencies is a major challenge for genetic studies, because available samples with sequencing data in AD are underpowered. Notably, almost all the genes with rare variants associated with AD risk also present common variants associated with AD risk (i.e., TREM2, SORL1, ABCA7, ABCA1, PLCγ2 and ADAM10)53. Third, gene–gene and gene–environment interactions have not yet been studied in detail. Hence, by increasing the GWAS sample size and improving imputation panels, conventional and (above all) more complex analyses will have more statistical power and should enable the characterization of associations with rare/structural variants. Lastly, higher-powered GWASs of multiancestry populations will be particularly welcome for characterizing potential new genetic risk factors, improving fine-mapping approaches and developing specific GRSs (because GRSs developed with European-ancestry populations are known to be less effective with other ancestries).
In conclusion, we have validated 33 previous loci, doubled the total number of genetic loci associated with the ADD risk, expanded our current knowledge of the pathophysiology of ADD, identified new opportunities for the development of GRSs and gene-specific treatments and opened up a pathway to translational genomics and personalized medicine.
Methods
Samples
All of our stage I meta-analysis samples came from the following consortia/datasets: EADB, GR@ACE, EADI, GERAD/PERADES, DemGene, Bonn, the Rotterdam study, the CCHS study, NxC and the UKBB. In the UKBB, individuals who did not report dementia or any family history of dementia were used as controls; the analysis included 2,447 diagnosed cases, 46,828 proxy cases of dementia and 338,440 controls. All individuals included in stage I are of European ancestry; demographic data on these case–control studies are summarized in Supplementary Table 1, and more detailed descriptions are available in the Supplementary Note. Stage II samples are from the ADGC, CHARGE and FinnGen consortia (Supplementary Table 1 and Supplementary Note) and are described in detail elsewhere5,6,9,10,54,55,56. Written informed consent was obtained from study participants or, for those with substantial cognitive impairment, a caregiver, legal guardian or other proxy. Study protocols for all cohorts were reviewed and approved by the appropriate institutional review boards.
Quality control and imputation
A standard quality control was performed on variants and samples from all datasets individually. The samples were then imputed with the TOPMed reference panel57,58. The Haplotype Reference Consortium (HRC) panel59 was also used for some datasets (Supplementary Table 2). For the UKBB, we used the provided imputed data generated from a combination of the 1000 Genomes, HRC and UK10K reference panels (Supplementary Note).
Stage I analyses
Tests of the association between clinical or proxy-ADD status and autosomal genetic variants were conducted separately in each dataset by using logistic regression and an additive genetic model, as implemented in SNPTEST 2.5.4-beta3 (ref. 60) or PLINK v1.90 (ref. 4). However, a logistic mixed model (as implemented in SAIGE v0.36.4 (ref. 61)) was considered for the UKBB data. We analyzed the genotype probabilities in SNPTEST (using the newml method) and dosages in PLINK and SAIGE. Analyses were adjusted for PCs and genotyping centers, when necessary (Supplementary Table 2). For the UKBB dataset, only variants with a MAF above 0.01% and a minor allele count (MAC) above 3 were analyzed, and effect sizes and standard errors were corrected by a factor of two, because proxy cases were analyzed7. This approach is appropriate for variants with a moderate-to-high frequency and a small effect size. For all datasets, we filtered out duplicated variants and variants with (1) missing data on the effect size, standard error or P value; (2) an absolute effect size above 5; (3) an imputation quality below 0.3; and (4) a value below 20 for the product of the MAC and the imputation quality (MAC-info score). For datasets not imputed with the TOPMed reference panel, we also excluded (1) variants for which conversion of position or alleles from the GRCh37 assembly to the GRCh38 assembly was not possible or problematic or (2) variants with very large difference of frequency between the TOPMed reference panel and the reference panels used to perform imputation.
Results were then combined across studies in a fixed-effect meta-analysis with an inverse-variance weighted approach, as implemented in METAL v2011-03-25 software62. We filtered out (1) variants with a heterogeneity P value below 5 × 10−8, (2) variants analyzed in less than 20% of the total number of cases and (3) variants with frequency amplitude above 0.4 (defined as the difference between the maximum and minimum frequencies across all the studies). We also excluded variants not analyzed in the EADB-TOPMed dataset.
The genomic inflation factor lambda was computed with the GenABEL 1.8-0 R package63 and a median approach after exclusion of the APOE region (44–46 Mb on chromosome 19 in GRCh38). The LD score regression intercept was computed with LDSC v1.0.1 software using the ‘baselineLD’ LD scores built from 1000 Genomes phase 3 (ref. 64). The analysis was restricted to HapMap 3 variants and excluded multiallelic variants, variants without an rs ID and variants in the APOE region.
Definition of associated loci
A region of ±500 kb was defined around each variant with a stage I P value below 1 × 10−5. These regions were then merged (using bedtools v2.27.0 software; https://bedtools.readthedocs.io/en/latest/) to define nonoverlapping regions. The region corresponding to the APOE locus was excluded. We then used the PLINK clumping procedure to define independent hits in each region. An iterative clumping procedure was applied to all variants with a stage I P value below 1 × 10−5, starting with the variant with the lowest P value (referred to as the index variant). Variants with a stage I P value below 1 × 10−5, located within 500 kb of this index variant and in LD with the index variant (r2 above 0.001) were assigned to the index variant’s clump. The clumping procedure was then applied until all the variants had been clumped. LD in the EADB-TOPMed dataset was computed using high-quality (probability ≥0.8) imputed genotypes.
Stage II analyses
Variants with a stage I P value below 1 × 10−5 were followed up (Supplementary Note). Results were combined across all stage I and II studies in a fixed-effect meta-analysis with an inverse variance weighted approach, as implemented in METAL. In each clump, we then reported the variants with positive follow-up results (i.e., the same direction of effect in stage I and stage II, and a stage II P value below 0.05) and the lowest P value in the meta-analysis. Those variants were considered to be associated at the genome-wide significance level if they had a P value below 5 × 10−8 in the stage I and II meta-analysis. However, we excluded the chr6:32657066:G:A variant, because its frequency amplitude was high.
Pathway analysis
A total of 10,271 gene sets were considered for analysis (Supplementary Note). Gene set enrichment analyses were performed in MAGMA v1.08 (refs. 65,66), with correction for the number of variants in each gene, LD between variants and LD between genes. LD was computed from the EADB-TOPMed dataset using high-quality (probability ≥0.9) imputed genotypes. The measure of pathway enrichment was the MAGMA ‘competitive’ test (in which the association statistic for genes in the pathway is compared with those for all other protein-coding genes), as recommended by De Leeuw et al.67. We used the ‘mean’ test statistic, which uses the sum of −log(variant P value) across all genes. The primary analysis assigned variants to genes if they lay within the gene boundaries, although a secondary analysis used a window of 35 kb upstream and 10 kb downstream to assign variants to genes (as in Kunkle et al.5). The primary analysis included all variants with an imputation quality above 0.8. We used q values68 to account for multiple testing.
Expression in various cell types
The expression of genes was assigned to specific cell classes of the adult brain, as described previously69. Briefly, middle temporal gyrus single-nucleus transcriptomes from the Allen Brain Atlas dataset (49,555 total nuclei derived from 8 human tissue donors aged 24–66 years) were used to annotate and select six main cell classes using Seurat 3.1.1 (ref. 70): glutamatergic neurons, GABAergic neurons, astrocytes, oligodendrocytes, microglia and endothelial cells. Enrichment analyses were performed by using the mean gene expression per nucleus for each cell type relative to the total expression summed across cell types as a quantitative covariate in a MAGMA gene property analysis.
Functional interpretation of GWAS signals and gene prioritization
To prioritize candidate genes in the new loci, we systematically searched for evidence for these genes in seven different domains: (1) variant annotation, (2) eQTL-GWAS integration, (3) sQTL-GWAS integration, (4) protein QTL (pQTL)-GWAS integration, (5) mQTL-GWAS integration, (6) histone acetylation QTL (haQTL)-GWAS integration and (7) APP metabolism. On the basis of this evidence, we then defined a gene prioritization score of between 0 and 100 for each candidate gene (Supplementary Fig. 34). Detailed information on the domains, categories (e.g., the tissue or cell type for QTL-GWAS integration domains) and subcategories (for the type of evidence) is given in Supplementary Table 19. A brief summary of how evidence was assessed in each domain is provided below, together with a detailed description of the gene prioritization strategy.
Candidate genes
We considered protein-coding candidate genes within a ±1-Mb window of the new lead variants. The genes in overlapping loci (i.e., L28, L30 and L37) were assigned to their respective loci based on proximity to the lead variants, and the distal genes were not considered for gene prioritization in the investigated loci. Moreover, we did not perform gene prioritization in the complex IGH gene cluster locus (L27), as this telomeric region contains complex splicing events (spanning a high number of IGH genes) that probably result from known fusion events18.
The variant annotation domain
In this domain, we determined whether the candidate gene was the nearest protein-coding gene to the lead variant and/or whether the lead variant was a rare variant (MAF < 1%) and/or protein-altering variant of the investigated candidate gene.
Molecular QTL–GWAS integration domains
To study the downstream effects of new ADD-associated variants on molecular phenotypes (i.e., expression, splicing, protein expression, methylation and histone acetylation) in various AD-relevant tissues, cell types and brain regions, molecular cis-QTL information (i.e., the genetic variants that regulate these molecular phenotypes) was integrated with the stage I ADD GWAS results in genetic colocalization analyses, TWASs and a genetically driven DNA methylation scan. These molecular QTLs include eQTLs, sQTLs, pQTLs, mQTLs and haQTLs. We mapped and prepared eQTL/sQTL catalogs in AD-relevant bulk brain regions from AMP-AD cohorts71,72,73,74 and in LCLs from the EADB Belgian cohort. We used additional eQTL/sQTL information in AD-relevant bulk brain regions from GTEx75 and microglia from the MiGA study76. Furthermore, eQTLs in monocytes and macrophages from various datasets77,78,79,80,81,82 (as prepared by eQTL Catalogue83) were included in the analyses. Data on pQTLs84, mQTLs85 and haQTLs85 were available for DLPFC. Using each molecular QTL catalogue, the effect of the lead variants was queried and significant associations were reported. Moreover, genetic colocalization studies were conducted by comparing ADD association signals with the eQTL/sQTL signals from AMP-AD bulk brain, MiGA microglia and EADB LCL cohorts. We also conducted eTWASs and splicing TWASs (sTWAS) of the ADD risk, along with fine mapping of the eTWAS results. To this end, we trained functional expression and splicing reference panels based on the AMP-AD bulk brain and EADB LCL cohorts, and we leveraged precalculated reference panel weights86 for the GTEx dataset75 in tissues and cells of interest. Lastly, for the mQTL-GWAS integration domain, we also tested for associations between ADD and genetically driven DNA methylation (MetaMeth analysis) in blood (with blood–brain methylation correlation estimates obtained from BECon87) using the procedures described by Freytag et al.88 and Barbeira et al.89. A detailed description of the datasets and methods used for each of these analyses is given in the Supplementary Note.
APP metabolism domain
We assessed the functional impact of gene underexpression on APP metabolism for all candidate genes based on a genome-wide high-content short interfering RNA screen17 (Supplementary Note).
Gene prioritization score
We computed a gene prioritization score for each candidate gene as the weighted sum of the evidence identified in the seven domains. We specified a weight for each type of evidence, as detailed in Supplementary Table 19. For the molecular QTL-GWAS integration domains, we gave more weight to replicated hits (i.e., evidence in several datasets) than to single hits. We also gave more weight to hits observed in brain (the bulk brain and microglia datasets) than to hits observed in other tissues/cell types (LCLs, monocytes, macrophages and blood). To avoid score inflation, several specific rules were applied: (1) for the results of sQTL- and mQTL-based analyses, multiple splice junctions or CpGs annotated for the same genes were aggregated prior to weighting due to correlated data; (2) if we observed a fine-mapped eTWAS association for a gene, its other significant (but not fine-mapped) eTWAS associations were not considered; (3) for genes having several significant CpGs (prior to aggregation) in MetaMeth analyses, the associated CpGs with a low (<75% percentile) blood–brain methylation correlation estimate were not considered if the gene also had associated CpGs with a high (≥75% percentile) blood–brain methylation correlation estimate.
Gene prioritization strategy
After obtaining a total weighted score per gene, we ranked genes per locus according to their prioritization scores and compared the relative score differences between the highest ranked gene and other genes in the investigated locus. If this relative difference was at least 20% and the gene prioritization score for the highest ranked gene was ≥4, then we classified this gene as a tier 1 prioritized gene in the investigated locus (i.e., a greater likelihood of being the true risk gene responsible for the ADD signal). If this absolute threshold was not met, then the highest ranked gene was classified as a tier 2 prioritized gene (i.e., a lower level of confidence and absence of the minimum level of evidence for a true risk gene). Furthermore, other genes in a locus harboring a tier 1 gene were classified as tier 2 prioritized genes if the relative score difference versus the highest ranked (tier 1) gene was between 20% and 50%. Lastly, when the relative score difference between the highest ranked gene and other genes in the same locus was <20%, then both the highest ranked gene and all genes with a score difference <20% were classified as tier 2 prioritized genes in the investigated locus; based on the current evidence, it is difficult to prioritize two or more similarly scored genes. The gene prioritization strategy is summarized in Supplementary Fig. 34. Detailed descriptions and discussions of prioritized genes and tier levels in each investigated new locus can be found in the Supplementary Note.
GRS analysis
Eight longitudinal MCI cohorts and seven population-based studies were included in the analysis and are fully described in the Supplementary Note and Supplementary Table 33. The GRSs were calculated as previously described90. Briefly, we considered variants with genome-wide significant evidence of association with ADD in our study. We did not include any APOE variants in the GRS. Variants were directly genotyped or imputed (R² ≥ 0.3). Imputation was performed using the HRC panel59 for subcohorts from the Rotterdam study and the TOPMed panel for the other cohorts57. For HRC-imputed data, LD proxies were considered for variants that were not available in this reference panel. The GRS was calculated as the weighted average of the number of risk-increasing alleles for each variant, using dosages. Weights were based on the respective log(OR) obtained in stage II. The GRS was then multiplied by the number of included variants. Thus, the HR measured the effect of carrying one additional average risk allele.
To assess whether the new variants in this study contribute to the risk of conversion to AD (in addition to known AD genes), we calculated two GRSs: one based solely on variants known before this study (GRSknown, n = 39; Table 1) and another based on variants identified in the present study (GRSnovel, n = 44; Table 2). These GRSs were calculated in the same way as the GRS encompassing all the variants.
The association between the GRS and the risk of progression to dementia in individuals from population-based cohorts or patients with MCI from memory clinics was tested statistically using Cox proportional hazards models. The models were adjusted for age, sex, the first four PCs (to correct for potential population stratification) and the number of APOE-ε4 and APOE- ε2 alleles (assuming an additive effect). In the FHS study, the generation was used as an additional covariate. In the 3C study, the analysis was adjusted for age, sex, the number of APOE alleles, the two first PCs and center. The PCs used were generated for each cohort, using the same variants as in the case/control study’s PC analysis. The number of APOE-ε4 alleles was obtained from direct genotyping or, if missing, the genotypes (with probability >0.8) derived from the TOPMed imputations. The interaction between the GRS and the number of APOE-ε4 alleles was tested on the multiplicative scale. In the primary analysis, conversion to AD was used as the outcome (conversions to non-AD dementias were coded as being censored at time of conversion), but analyses were repeated using all-cause dementia as the outcome.
To quantify the effect size of the potential association between the GRS and conversion to dementia regarding predictive performance, we computed three different indices measuring different aspects of the predictive performance of the GRS in our prospective, longitudinal cohort studies91: the continuous version of the C-index,92,93 the continuous NRI94 and IPA95 (Supplementary Note). For all indices, we provide point estimates and 95% CIs.
In the main analysis, indices were computed at the time point for which all cohorts in a specific setting (i.e., population-based studies or memory clinics, respectively) provided follow-up observations (that is 5 years for population-based cohorts and 3 years for MCI cohorts). In a sensitivity analysis, indices for longer or shorter follow-up periods were also derived (that is 3 years and 10 years for population-based cohorts and 5 years for MCI cohorts). Standard errors for indices were derived by non-parametric bootstrapping with 1,000 samples.
To determine the average effect of the GRS across the various cohorts examined, individual cohort results were subjected to both inverse-variance weighted meta-analyses (primary analyses) and random effects meta-analysis (Supplementary Note). To facilitate comparisons of results for different time points, cohorts with longer follow-up periods were meta-analyzed separately. Furthermore, two memory clinic cohorts with a limited sample size (N < 50) were excluded to assess their impact on the final meta-analysis results. Meta-analyses were performed using the ‘metafor’ (3.0.2) R package96.
To further illustrate the clinical relevance of the GRS, we pooled computed GRSs across four population-based cohorts (3C, AgeCoDe, VITA and MAS) and computed deciles of the GRS distribution for use as a common reference for all cohorts. We then computed the increase in risk when augmenting the GRS value from the first decile (GRS = 50.76) to the ninth decile (GRS = 59.74) of the distribution. To represent this risk increase in the HR, we rescaled the HR derived from our meta-analyses results using the equation \(e^{\log\left( {\rm HR} \right) \ast \left( {{\rm{GRS9th}}_{\rm{decile}}} - {{\rm{GRS1st}}_{\rm{decile}}} \right)}\). Importantly, this approach yields exactly the same results as transforming the GRS so that a one unit increment corresponds to the increase from the lowest decile to the highest decile.
Furthermore, we approximated the probability of conversion to AD at 3 and 5 years in memory clinic patients with MCI by using Cox models implemented in the ‘PredictCox’ function from the ‘riskRegression’ (2020.12.8) R package97. We did not derive AD conversion probabilities for two cohorts with very small sample sizes (N < 50). Predicted AD conversion probabilities were derived and averaged for all patients in each of the groups formed by the decile of the GRS distribution in each cohort. The difference between the groups with the highest and lowest GRSs was computed in each cohort. We report the median (range) results in each group formed by the GRS deciles.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data Availability
Genome-wide summary statistics have been deposited to the European Bioinformatics Institute GWAS Catalog (https://www.ebi.ac.uk/gwas/) under accession no. GCST90027158.
The significant eQTLs/sQTLs mapped and eTWAS/sTWAS functional reference panel weights generated for this study (in AD-relevant bulk brain regions from AMP-AD cohorts and in LCLs from the EADB Belgian cohort) are publicly available at https://doi.org/10.5281/zenodo.5745927 and https://doi.org/10.5281/zenodo.5745929.
Anonymized aligned reads of the amplicon-based long-read Nanopore cDNA sequencing experiment conducted for the TSPAN14 splicing analysis are available through the European Nucleotide Archive under accession PRJEB49234.
Moreover, the following data used in the gene prioritization are publicly available:
AMP-AD rnaSeqReprocessing Study (https://www.synapse.org/#!Synapse:syn9702085);
MayoRNAseq whole-genome sequencing variant call formats (WGS VCFs) (https://www.synapse.org/#!Synapse:syn11724002);
ROSMAP WGS VCFs (https://www.synapse.org/#!Synapse:syn11724057);
MSBB WGS VCFs (https://www.synapse.org/#!Synapse:syn11723899);
eQTLGen (https://www.eqtlgen.org/);
eQTL Catalogue database (https://www.ebi.ac.uk/eqtl/);
Brain xQTL serve (http://mostafavilab.stat.ubc.ca/xqtl/);
GTEx v8 eQTL and sQTL catalogs (https://www.gtexportal.org/);
GTEx v8 expression and splicing prediction models (http://predictdb.org/);
MiGA eQTLs (https://doi.org/10.5281/zenodo.4118605);
MiGA sQTLs (https://doi.org/10.5281/zenodo.4118403);
MiGA meta-analysis (https://doi.org/10.5281/zenodo.4118676); and
Wingo et al.84 pQTL data (https://www.synapse.org/#!Synapse:syn23627957).
Code availability
We used publicly available software for all analyses. The software are listed in the Supplementary Note with their appropriate citations and/or URLs.
References
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Bellenguez, C., Grenier-Boley, B. & Lambert, J. C. Genetics of Alzheimer’s disease: where we are, and where we are going. Curr. Opin. Neurobiol. 61, 40–48 (2020).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Liu, J. Z., Erlich, Y. & Pickrell, J. K. Case-control association mapping by proxy using family history of disease. Nat. Genet. 49, 325–331 (2017).
Marioni, R. E. et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99 (2018).
Sims, R. et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease.Nat. Genet. 49, 1373–1384 (2017).
Jun, G. et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117 (2016).
Schwartzentruber, J. et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 53, 392–402 (2021).
de Rojas, I. et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat. Commun. 12, 3417 (2021).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Skene, N. G. et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 50, 825–833 (2018).
de Leeuw, C. A., Stringer, S., Dekkers, I. A., Heskes, T. & Posthuma, D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat. Commun. 9, 3768 (2018).
Haass, C., Kaether, C., Thinakaran, G. & Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270 (2012).
Chapuis, J. et al. Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. 133, 955–966 (2017).
Cleynen, A. et al. Expressed fusion gene landscape and its impact in multiple myeloma. Nat. Commun. 8, 1893 (2017).
Szklarczyk, D. et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Spit, M., Rieser, E. & Walczak, H. Linear ubiquitination at a glance. J. Cell Sci. 132, jcs208512 (2019).
Pencina, M. J., D’Agostino, R. B., Pencina, K. M., Janssens, A. C. J. W. & Greenland, P. Interpreting incremental value of markers added to risk prediction models. Am. J. Epidemiol. 176, 473–481 (2012).
Dourlen, P., Chapuis, J. & Lambert, J.-C. Using high-throughput animal or cell-based models to functionally characterize GWAS signals. Curr. Genet. Med. Rep. 6, 107–115 (2018).
Dourlen, P., Kilinc, D., Malmanche, N., Chapuis, J. & Lambert, J. C. The new genetic landscape of Alzheimer’s disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 138, 221–236 (2019).
Deuss, M., Reiss, K. & Hartmann, D. Part-time α-secretases: the functional biology of ADAM 9, 10 and 17. Curr. Alzheimer Res. 5, 187–201 (2008).
Kim, T. et al. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404 (2013).
Salminen, A. & Kaarniranta, K. Siglec receptors and hiding plaques in Alzheimer’s disease. J. Mol. Med. 87, 697–701 (2009).
Rodgers, M. A. et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 211, 1333–1347 (2014).
Iwai, K. LUBAC-mediated linear ubiquitination: a crucial regulator of immune signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 97, 120–133 (2021).
Venegas, C. et al. Microglia-derived ASC specks crossseed amyloid-β in Alzheimer’s disease. Nature 552, 355–361 (2017).
Ising, C. et al. NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673 (2019).
Nakayama, Y. et al. Linear polyubiquitin chain modification of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 79, 256–265 (2020).
Black, R. A. et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-∅ from cells. Nature 385, 729–733 (1997).
Verstrepen, L., Carpentier, I., Verhelst, K. & Beyaert, R. ABINs: A20 binding inhibitors of NF-κB and apoptosis signaling. Biochem. Pharmacol. 78, 105–114 (2009).
Spitz, C. et al. Non-canonical Shedding of TNFα by SPPL2a is determined by the conformational flexibility of its transmembrane helix. iScience 23, 101775 (2020).
Tang, W. et al. The growth factor progranulin binds to tnf receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484 (2011).
He, P. et al. Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 178, 829–841 (2007).
Shi, J. Q. et al. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 1368, 239–247 (2011).
Bezbradica, J. S., Coll, R. C. & Schroder, K. Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell. Mol. Immunol. 14, 118–126 (2017).
Decourt, B., Lahiri, D. K. & Sabbagh, M. N. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr. Alzheimer Res. 14, 412–425 (2016).
De Strooper, B. & Karran, E. The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016).
Gong, K. et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J. Clin. Invest. 128, 2500–2518 (2018).
Rhinn, H. & Abeliovich, A. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst. 4, 404–415 (2017).
Vass, R. et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 121, 373–380 (2011).
Baizabal-Carvallo, J. F. & Jankovic, J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 12, 175–185 (2016).
Tropea, T. F. et al. TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann. Neurol. 85, 801–811 (2019).
Mendsaikhan, A., Tooyama, I. & Walker, D. G. Microglial progranulin: involvement in Alzheimer’s disease and neurodegenerative diseases. Cells 8, 230 (2019).
Li, Z. et al. The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol. 139, 45–61 (2020).
Yang, H. S. et al. Genetics of gene expression in the aging human brain reveal TDP-43 proteinopathy pathophysiology. Neuron 107, 496–508.e6 (2020).
Paushter, D. H., Du, H., Feng, T. & Hu, F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 136, 1–17 (2018).
Feng, T., Lacrampe, A. & Hu, F. Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol. 141, 327–339 (2021).
Lacour, A. et al. Genome-wide significant risk factors for Alzheimer’s disease: role in progression to dementia due to Alzheimer’s disease among subjects with mild cognitive impairment. Mol. Psychiatry 22, 153–160 (2017).
Zhang, Q. et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 11, 1–11 (2020).
Holstege, H. et al. Exome sequencing identifies novel AD-associated genes. medRxiv 18, 24 (2020).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circulation: Cardiovasc. Genet. 2, 73–80 (2009).
Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441 (2011).
Jun, G. et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 67, 1473–1484 (2010).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913 (2007).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Aulchenko, Y. S., Ripke, S., Isaacs, A. & van Duijn, C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007).
Bulik-Sullivan, B. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Yurko, R., Roeder, K., Devlin, B. & G’Sell, M. H‐MAGMA, inheriting a shaky statistical foundation, yields excess false positives. Ann. Hum. Genet. 85, 97–100 (2021).
De Leeuw, C. A., Neale, B. M., Heskes, T. & Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 17, 353–364 (2016).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. U. S. A. 100, 9440–9445 (2003).
Coelho, D. M., L. I. da Cruz Carvalho, Melo-de-Farias, A. R., Lambert, J.-C. & Costa, M. R. Differential transcript usage unravels gene expression alterations in Alzheimer’s disease human brains. NPJ Aging Mech. Dis. 7, 2 (2021).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Allen, M. et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089 (2016).
Mostafavi, S. et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 21, 811–819 (2018).
Bennett, D. A. et al. Religious orders study and rush memory and aging project. J. Alzheimer’s Dis. 64, S161–S189 (2018).
Wang, M. et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci. Data 5, 180185 (2018).
GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
De, K. et al. Atlas of genetic effects in human microglia transcriptome across brain regions, aging and disease pathologies. Preprint at bioRxiv https://doi.org/10.1101/2020.10.27.356113 (2020).
Alasoo, K. et al. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 50, 424–431 (2018).
Nédélec, Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–669.e21 (2016).
Chen, L. et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414.e24 (2016).
Momozawa, Y. et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat. Commun. 9, 2427 (2018).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Sci. (80-.). 343, 1246949 (2014).
Quach, H. et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell 167, 643–656.e17 (2016).
Kerimov, N. et al. eQTL catalogue: a compendium of uniformly processed human gene expression and splicing QTLs. Preprint at bioRxiv https://doi.org/10.1101/2020.01.29.924266 (2020).
Wingo, A. P. et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat. Genet. 53, 143–146 (2021).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Barbeira, A. N. et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 22, 49 (2021).
Edgar, R. D., Jones, M. J., Meaney, M. J., Turecki, G. & Kobor, M. S. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 7, e1187 (2017).
Freytag, V. et al. Genetic estimators of DNA methylation provide insights into the molecular basis of polygenic traits. Transl. Psychiatry 8, 31 (2018).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Chouraki, V. et al. Evaluation of a genetic risk score to improve risk prediction for Alzheimer’s disease. J. Alzheimers Dis. 53, 921–932 (2016).
Steyerberg, E. W. et al. Assessing the performance of prediction models: a framework for traditional and novel measures. Epidemiology 21, 128–138 (2010).
Harrell, F. E., Califf, R. M., Pryor, D. B., Lee, K. L. & Rosati, R. A. Evaluating the yield of medical tests. JAMA 247, 2543–2546 (1982).
Gerds, T. A., Kattan, M. W., Schumacher, M. & Yu, C. Estimating a time-dependentconcordance index for survival prediction models with covariate dependent censoring. Stat. Med. 32, 2173–2184 (2013).
Pencina, M. J., D’Agostino, R. B. & Steyerberg, E. W. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat. Med. 30, 11–21 (2011).
Kattan, M. W. & Gerds, T. A. The index of prediction accuracy: an intuitive measure useful for evaluating risk prediction models. Diagnostic Progn. Res. 2, 7 (2018).
Viechtbauer, W. Conducting meta-analyses in R with the metafor. J. Stat. Softw. 36, 1–48 (2010).
Ozenne, B., Sørensen, A. L., Scheike, T., Torp-Pedersen, C. & Gerds, T. riskRegression: predicting the risk of an event using Cox regression models. R. J. 9, 440–460 (2017).
Acknowledgements
We thank the many study participants, researchers and staff for collecting and contributing to the data, the high-performance computing service at the University of Lille and the staff at CEA-CNRGH for their help with sample preparation and genotyping and excellent technical assistance. We thank Antonio Pardinas for his help. We thank the Netherlands Brain Bank. This research was conducted using the UKBB resource (application number 61054). This work was funded by a grant (EADB) from the EU Joint Programme – Neurodegenerative Disease Research. INSERM UMR1167 is also funded by the INSERM, Institut Pasteur de Lille, Lille Métropole Communauté Urbaine and French government’s LABEX DISTALZ program (development of innovative strategies for a transdisciplinary approach to AD). Full consortium acknowledgements and funding are in the Supplementary Note.
Author information
Authors and Affiliations
Consortia
Contributions
EADB coordination: K. Mather, F.J., M.T., R.F.-S., J. Clarimon, J.-F. Deleuze, O.A.A., M.I., M. Hiltunen, K.S., C.M.v.D., R.S., W.M.v.d.F., A. Ruiz, A. Ramirez and J.-C.L. Data analyses: C. Bellenguez, F.K., I.E.J., L.K., S.M.-G., N.A., R.C., B.G.-B., V. Andrade, P.A.H., R.C.-M., V.D., S.J.v.d.L., M.R.C., T.K., I.R., J. Chapuis and P.G.-G. ADGC analysis and coordination: A.C.N., W.S.B., L.A.F., J.L.H., K.L.H.-N., P.P.K., B.W.K., C.-Y.L. and E.R.M., R. Mayeux, M.A.P.-V., J.S., L.-S.W., Y.Z. and G.D.S. Charge analysis and coordination: Q.Y., J.C.B., A.D.S., C.S., B.M.P., R.W., O. Lopez. and S. Seshadri. FinnGen analysis: T.K. and M. Hiltunen. Rotterdam analysis: A.Y., I.P.-N, M. Ghanbari and M.A.I. Sample contribution: S. Ahmad, V. Giedraitis, D. Aarsland, P.V.Ã., D.G.-G., C. Abdelnour, E.A.-M., D. Alcolea, M. Alegret, I. Alvarez, V. Alvarez, N.J.A., A. Tsolaki, C. Antúnez, I. Appollonio, M. Arcaro, S. Archetti, A.A.P., B.A., L.A., H. Bailly, N.B., M. Baquero, S. Barral, A. Beiser, A.B.P., J.E.B., P. Benchek, L.B., C. Berr, C. Besse, V. Bessi, G. Binetti, A. Bizarro, R.B., M. Boada, E.B., B.B., S. Boschi, P. Bossù, G. Bråthen, J.B., C. Bresner, H. Brodaty, K.J.B., L.I.B., D.B.-R., K.B., V. Burholt, W.S.B., M.C., L.B.C., G.C., J. Chung, M.L.C., Ã.C., R.C., L.C.-C., C. Charbonnier, H.-H.C., C. Chilotti, S.C., J.A.C., C. Clark, E. Conti, A.C.-G., E. Costantini, C. Custodero, D.D., M.C.D., A. Daniele, E. Dardiotis, J-F. Dartigues, P.P.d.D., K.d.P.L., L.D.d.W., S. Debette, J.D., T.d.S., N.D., A. DeStefano, M.D., J.D.-S., M.D.-F., P.D.R., S. Djurovic, E. Duron, E. Duzel, C.D., G.E., S.E., V.E.-P., A.E., M.E., K.M.F., T. Fabrizio, S.F.N., D.W.F., L. Farotti, C.F., M.F.-F., R.F., C.B.F., E.F., B. Fin, P.F., T. Fladby, K.F., B. Fongang, M.F., J.F., T.M.F., S.F., N.C.F., E.F.-M., M.J.B., A.F.-G., L. Froelich, B.F.-H., D.G., J.M.G.-A., S.G.-M., G.G.-R., R.G., I.G., G. Giorgio, A.M.G., O.G., D.G.-F., A.G.-P., C.G., G. Grande, E. Green, T.G., E. Grunblatt, M. Grunin, V. Gudnason, T.G.-B., A.H., G.H., J.L.H., K.L.H.-N., H. Hampel, O.H., J. Hardy, A.M.H., L.H., J. Harwood, S.H.-H., S.H., M.T.H., I.H., M.J.H., P.H., C.H., H. Holstege, R.H.V., M. Hulsman, J. Humphrey, G.J.B., X.J., C.J., G.R.J., Y.K., J. Kauwe, P.G.K., L. Kilander, A.K.S., M.K., A.K., J. Kornhuber, M.H.K., W.A.K., P.P.K., B.W.K., A.B.K., C.L., E.J.L., L. Launer, A. Lauria, C.-Y.L., J.L., O.Ler., A. Lleó, W.L.J., O. Lopez, A.L.d.M., S.L., M.L., L. Luckcuck, K.L.L., Y.M., J.M., C.A.M., W.M., F. Mangialasche, M. Spallazzi, M. Marquié, R. Marshall, E.R.M., A.M.M., C.M.R., C. Masullo, R. Mayeux, S. Mead, P. Mecocci, M. Menéndez-González, A.M., S. Mehrabian, S. Mendoza, M.M.-G., P. Mir, S. Moebus, M. Mol, L.M.-P., L. Montrreal, L. Morelli, F. Moreno, K. Morgan, T. Mosley, M.M.N., C. Muchnik, S. Mukherjee, B.N., T.N., G.N., B.G.N., R.O., A.O., M.O., G.O., A.P., C. Paollo., G. Papenberg, L.P., F.P., P. Pastor, G. Peloso, A.P.-C., J.P.-T., P. Pericard, O.P., Y.A.P., J.A.P., G.P.-R., C. Pisanu, T.P., J. Popp, D.P., J. Priller, R.P., O.Q., I.Q., J.Q.T., A. Rábano, I. Rainero, F.R., I. Ramakers, L.M.R., M.J.R., C.R., D.R.-D., P. Ridge, S.R.-H., P. Riederer, N.R., E.R.-R., A. Rongve, I.R.A., M.R.-R., J.L.R., E.R., D.R., M.E.S., P. Sakka, I.S., Ã.S., M.B.S.-A., F.S.-G., P.S.J., R.S.-V., S.B.S., C.S., C.L.S., M. Scamosci, N. Scarmeas, E. Scarpini, P. Scheltens, N. Scherbaum, M. Scherer, M. Schmid, A. Schneider, J.M.S., G. Selbæk, D.S., M. Serrano, J.S., A.A.S., O.S., S. Slifer, G.J.L.S., H.S., V.S., A. Solomon, Y.S., S. Sorbi, O.S.-G., G. Spalletta, A. Spottke, A. Squassina, E. Stordal, J.P.T., L. Tárraga, N.T., A. Thalamuthu, T.T., G.T., L. Traykov, L. Tremolizzo, A.T.-H., A. Uitterlinden, A. Ullgren, I.U., S.V., O.V., C.V.B., J. Vance, B.N.V., A.v.d.L., J.V.D., J.v.R., J.v.S., R.V., F.V., J.-S.V., J. Vogelgsang, M.V., M.W., D.W., L.-S.W., R.W., L.W., J. Wiltfnag, G.W., B.W., M.Y., H.Z., Y.Z., X.Z., C.Z., M.Z., L.A.F., B.M.P., M. Ghanbari, T.R., P. Sachdev, K. Mather, F.J., M.A.I., A.d.M., J. Hort, M.T. and M.A.P.-V. Core writing group: C. Bellenguez, F.K., V. Andrade, B.G.-B., P.A.H., R.C.-M., L.K., S.J.v.d.L., K.S., A. Ruiz, A. Ramirez and J.-C.L.
Corresponding authors
Ethics declarations
Competing interests
H. Hampel is an employee of Eisai. The present article was initiated and prepared as part of his academic position at Sorbonne University (Paris, France), and it reflects entirely and exclusively his own opinion. He serves as Senior Associate Editor for the Alzheimers & Dementia journal and has not received any fees or honoraria since May 2019. Before May 2019, H. Hampel received lecture fees from Servier, Biogen and Roche; research grants from Pfizer, Avid and MSD Avenir (paid to the institution); travel funding from Eisai, Functional Neuromodulation, Axovant, Eli Lilly and Company, Takeda, Zinfandel Pharmaceuticals, GE Healthcare and Oryzon Genomics; and consultancy fees from Qynapse, Jung Diagnostics, Cytox, Axovant, Anavex, Takeda, Zinfandel Pharmaceuticals, GE Healthcare, Oryzon Genomics and Functional Neuromodulation. He served as a scientific advisory board member for Functional Neuromodulation, Axovant, Eisai, Eli Lilly and Company, Cytox, GE Healthcare, Takeda and Zinfandel, Oryzon Genomics and Roche Diagnostics. The remaining authors declare no competing interests
Peer review
Peer review file
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note, Methods, Results and Figures 1–50.
Supplementary Tables
Supplementary Tables 1–47.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bellenguez, C., Küçükali, F., Jansen, I.E. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54, 412–436 (2022). https://doi.org/10.1038/s41588-022-01024-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-022-01024-z
This article is cited by
-
FORGEdb: a tool for identifying candidate functional variants and uncovering target genes and mechanisms for complex diseases
Genome Biology (2024)
-
Identifying flaws in the GWAS datasets of a published Mendelian randomization study: complementary re-evaluation and suggestion for analytical refinements
Journal of Translational Medicine (2024)
-
Unraveling the intercellular communication disruption and key pathways in Alzheimer’s disease: an integrative study of single-nucleus transcriptomes and genetic association
Alzheimer's Research & Therapy (2024)
-
Association of long-term exposure to various ambient air pollutants, lifestyle, and genetic predisposition with incident cognitive impairment and dementia
BMC Public Health (2024)
-
Gut microbiota-host lipid crosstalk in Alzheimer’s disease: implications for disease progression and therapeutics
Molecular Neurodegeneration (2024)
